引用本文: 王莎莎, 劉維鋒. Stickler綜合征一例. 中華眼底病雜志, 2018, 34(6): 598-599. doi: 10.3760/cma.j.issn.1005-1015.2018.06.016 復制
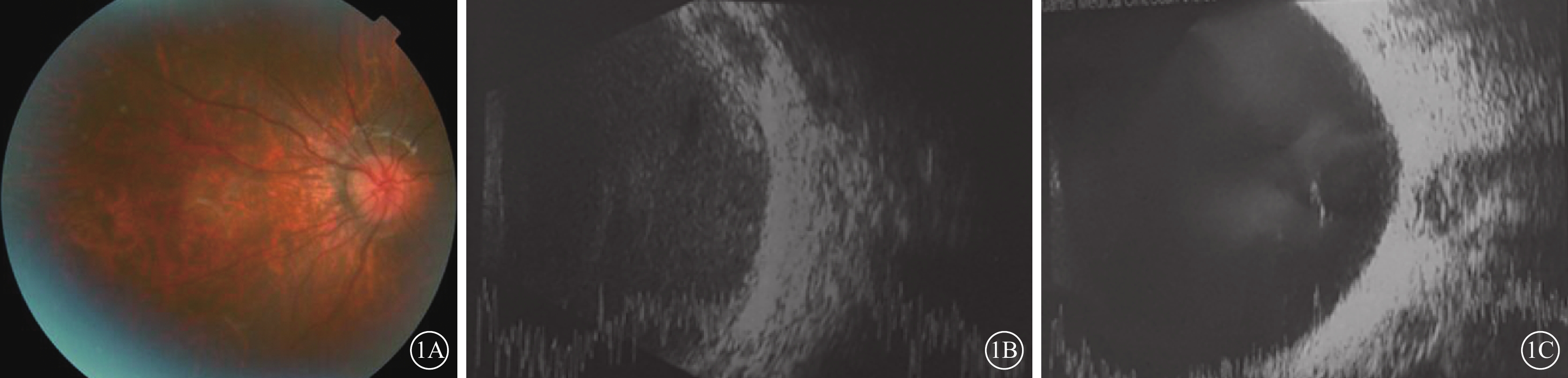
患兒女,7歲。因左眼視力下降6月余2017年10月到我院就診。患兒足月順產,父母非近親結婚,家族中無類似遺傳病史。2015年于外院診斷為“雙眼先天性白內障”并行雙眼白內障超聲乳化聯合人工晶狀體植入手術。全身檢查:患兒智力發育正常,顏面部小頜畸形改變,左右基本對稱,張口型正常,乳牙裂,牙槽突完整,雙側硬腭部分裂開,軟腭至懸雍垂完全裂開,舌體居中、活動自如,開口度及開口型無異常。心、肺、腹部檢查未見明顯異常,生理反射存在,病理反射未引出。眼部檢查:右眼視力0.04,矯正?8.50 DS/?2.00 DC×120°>0.3;左眼視力眼前手動,矯正無提高。雙眼眼前節正常,人工晶狀體位正。右眼玻璃體輕度混濁,眼底視盤邊界清楚、顏色淡,血管纖細,豹紋狀改變(圖1A);左眼玻璃體灰白混濁,眼底未窺及。右眼、左眼眼壓分別為21.7、7.3 mmHg(1 mmHg=0.133 kPa)。眼部B型超聲檢查,右眼玻璃體腔見纖細光帶漂浮;左眼玻璃體腔見V形強反射條帶,后運動試驗(?),玻璃體后脫離,玻璃體腔大量中回聲絮狀漂浮物,玻璃體后部見視網膜光帶結構紊亂、厚度不均,其下方大量機化膜且僵硬皺縮(圖1B,1C)。眼部A型超聲檢查,右眼眼軸長度30.47 mm,水平、垂直角膜曲率分別為48.21、46.49 DS;左眼無法測量。心臟彩色超聲檢查,三尖瓣輕度反流。純音聽閾測定提示雙耳輕度混合型耳聾。診斷:Stickler綜合征、左眼陳舊性視網膜脫離(漏斗狀)、雙眼人工晶狀體眼。建議右眼佩戴眼鏡矯正視力,弱視訓練;行腭裂修補手術。建議患者行基因檢測,但家長拒絕此項檢查。
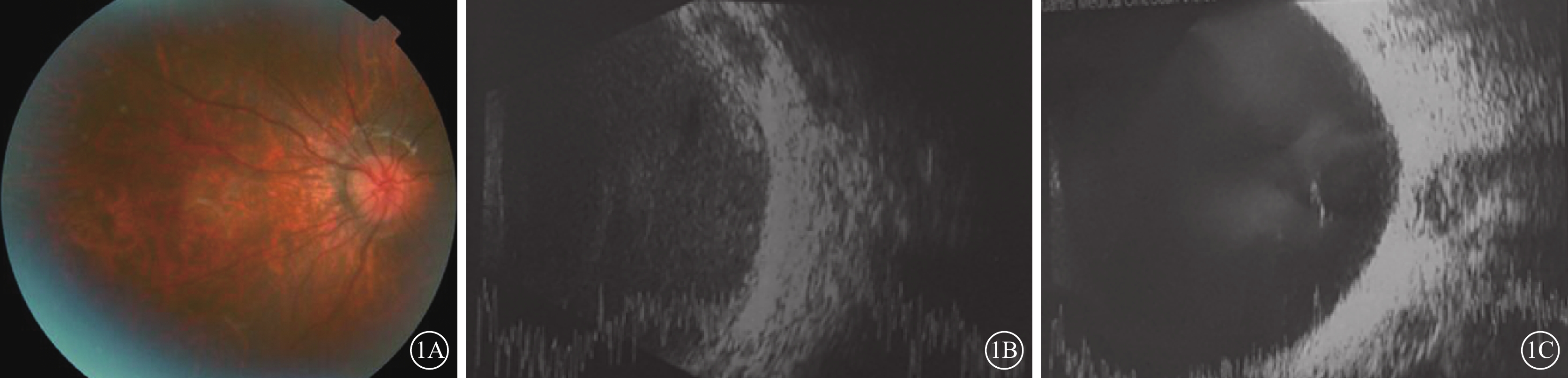 圖1
患兒彩色眼底像及眼部B型超聲像。1A.右眼彩色眼底像,視盤邊界清楚、顏色淡,血管纖細,視網膜豹紋狀改變;1B.右眼B型超聲像,玻璃體腔見纖細光帶漂浮;1C.左眼B型超聲像,玻璃體后脫離,玻璃體腔大量中回聲絮狀漂浮物,玻璃體后部見視網膜光帶結構紊亂、厚度不均,其下方見大量機化增生條索且僵硬皺縮
圖1
患兒彩色眼底像及眼部B型超聲像。1A.右眼彩色眼底像,視盤邊界清楚、顏色淡,血管纖細,視網膜豹紋狀改變;1B.右眼B型超聲像,玻璃體腔見纖細光帶漂浮;1C.左眼B型超聲像,玻璃體后脫離,玻璃體腔大量中回聲絮狀漂浮物,玻璃體后部見視網膜光帶結構紊亂、厚度不均,其下方見大量機化增生條索且僵硬皺縮
討論 Stickler綜合征是一種在臨床上較為少見的遺傳性進行性全身膠原結締組織病變,主要是由膠原蛋白的基因突變引起,導致全身廣泛的膠原蛋白功能紊亂而引起玻璃體、晶狀體、鞏膜及骨骼、關節、血管等一系列病變。盡管較多研究稱其大部分為常染色體顯性遺傳性疾病,但有50%的患兒是首發者[1-2]。本例患兒即為家族首發者。Stickler綜合征的特點是早期出現進行性近視、先天性白內障、青光眼、玻璃體退行性變、難治性視網膜脫離、腭裂、聽力下降、骨關節功能下降及運動障礙等。本例患兒7歲,特殊面容又伴有腭裂、先天性白內障、軸性高度近視、視網膜脫離伴聽力缺失,符合Stickler綜合征的診斷。
Stickler綜合征需與Wagner綜合征、Marshall綜合征以及侵蝕性玻璃體視網膜病變(ERVR)相鑒別。Stickler綜合征一般有玻璃體特征性改變(膜型或念珠型),而Wagner綜合征的特點為視網膜色素上皮病變、暗適應減退、視野缺損,其病變基礎是發生于染色體5q14.3上的基因突變[3]。Stickler綜合征一般存在較復雜的視網膜脫離情況,高度近視、暗適應常常為正常范圍,而Marshall綜合征臨床表現為白內障、低度近視、玻璃體異常、暗適應異常、較少發生視網膜脫離。并且,有學者在Marshall綜合征患者中發現了Ⅱ型膠原蛋白[4]。ERVR與Wagner綜合征的臨床癥狀非常相似,還可有視網膜半透明或視網膜侵蝕的表現;此外,其孔源性視網膜脫離的發病率也大大高于Wagner綜合征(73%比18%)[3]。
通過對不同基因位點突變的檢測,Stickler綜合征可分為3型。Ⅰ型被稱作遺傳性進行性骨關節眼病,是由于12號染色體上的COL2A1基因突變引起,此類型主要表現為眼、骨骼、聽力不同程度障礙[5]。Ⅱ型是由于第6號染色體上的COL11A2基因突變引起,在Ⅱ型原膠原基因三倍體螺旋線的小缺損可以導致骨骼發育異常,此類型主要導致骨發育異常而無眼部異常[6]。Ⅲ型是由第6號染色體上的COL11A1基因突變引起,主要伴有胎兒軟骨發育不良而無眼部異常[7],亦有人認為該型是Stickler綜合征的嬰兒型。基因檢測能確診該病,并能明確其分型以幫助治療[8]。但遺憾本例患兒家長拒絕行基因檢測。在缺少基因檢測的情況下該病與其他多種疾病的鑒別診斷尤為重要。
患兒女,7歲。因左眼視力下降6月余2017年10月到我院就診。患兒足月順產,父母非近親結婚,家族中無類似遺傳病史。2015年于外院診斷為“雙眼先天性白內障”并行雙眼白內障超聲乳化聯合人工晶狀體植入手術。全身檢查:患兒智力發育正常,顏面部小頜畸形改變,左右基本對稱,張口型正常,乳牙裂,牙槽突完整,雙側硬腭部分裂開,軟腭至懸雍垂完全裂開,舌體居中、活動自如,開口度及開口型無異常。心、肺、腹部檢查未見明顯異常,生理反射存在,病理反射未引出。眼部檢查:右眼視力0.04,矯正?8.50 DS/?2.00 DC×120°>0.3;左眼視力眼前手動,矯正無提高。雙眼眼前節正常,人工晶狀體位正。右眼玻璃體輕度混濁,眼底視盤邊界清楚、顏色淡,血管纖細,豹紋狀改變(圖1A);左眼玻璃體灰白混濁,眼底未窺及。右眼、左眼眼壓分別為21.7、7.3 mmHg(1 mmHg=0.133 kPa)。眼部B型超聲檢查,右眼玻璃體腔見纖細光帶漂浮;左眼玻璃體腔見V形強反射條帶,后運動試驗(?),玻璃體后脫離,玻璃體腔大量中回聲絮狀漂浮物,玻璃體后部見視網膜光帶結構紊亂、厚度不均,其下方大量機化膜且僵硬皺縮(圖1B,1C)。眼部A型超聲檢查,右眼眼軸長度30.47 mm,水平、垂直角膜曲率分別為48.21、46.49 DS;左眼無法測量。心臟彩色超聲檢查,三尖瓣輕度反流。純音聽閾測定提示雙耳輕度混合型耳聾。診斷:Stickler綜合征、左眼陳舊性視網膜脫離(漏斗狀)、雙眼人工晶狀體眼。建議右眼佩戴眼鏡矯正視力,弱視訓練;行腭裂修補手術。建議患者行基因檢測,但家長拒絕此項檢查。
圖1
患兒彩色眼底像及眼部B型超聲像。1A.右眼彩色眼底像,視盤邊界清楚、顏色淡,血管纖細,視網膜豹紋狀改變;1B.右眼B型超聲像,玻璃體腔見纖細光帶漂浮;1C.左眼B型超聲像,玻璃體后脫離,玻璃體腔大量中回聲絮狀漂浮物,玻璃體后部見視網膜光帶結構紊亂、厚度不均,其下方見大量機化增生條索且僵硬皺縮
討論 Stickler綜合征是一種在臨床上較為少見的遺傳性進行性全身膠原結締組織病變,主要是由膠原蛋白的基因突變引起,導致全身廣泛的膠原蛋白功能紊亂而引起玻璃體、晶狀體、鞏膜及骨骼、關節、血管等一系列病變。盡管較多研究稱其大部分為常染色體顯性遺傳性疾病,但有50%的患兒是首發者[1-2]。本例患兒即為家族首發者。Stickler綜合征的特點是早期出現進行性近視、先天性白內障、青光眼、玻璃體退行性變、難治性視網膜脫離、腭裂、聽力下降、骨關節功能下降及運動障礙等。本例患兒7歲,特殊面容又伴有腭裂、先天性白內障、軸性高度近視、視網膜脫離伴聽力缺失,符合Stickler綜合征的診斷。
Stickler綜合征需與Wagner綜合征、Marshall綜合征以及侵蝕性玻璃體視網膜病變(ERVR)相鑒別。Stickler綜合征一般有玻璃體特征性改變(膜型或念珠型),而Wagner綜合征的特點為視網膜色素上皮病變、暗適應減退、視野缺損,其病變基礎是發生于染色體5q14.3上的基因突變[3]。Stickler綜合征一般存在較復雜的視網膜脫離情況,高度近視、暗適應常常為正常范圍,而Marshall綜合征臨床表現為白內障、低度近視、玻璃體異常、暗適應異常、較少發生視網膜脫離。并且,有學者在Marshall綜合征患者中發現了Ⅱ型膠原蛋白[4]。ERVR與Wagner綜合征的臨床癥狀非常相似,還可有視網膜半透明或視網膜侵蝕的表現;此外,其孔源性視網膜脫離的發病率也大大高于Wagner綜合征(73%比18%)[3]。
通過對不同基因位點突變的檢測,Stickler綜合征可分為3型。Ⅰ型被稱作遺傳性進行性骨關節眼病,是由于12號染色體上的COL2A1基因突變引起,此類型主要表現為眼、骨骼、聽力不同程度障礙[5]。Ⅱ型是由于第6號染色體上的COL11A2基因突變引起,在Ⅱ型原膠原基因三倍體螺旋線的小缺損可以導致骨骼發育異常,此類型主要導致骨發育異常而無眼部異常[6]。Ⅲ型是由第6號染色體上的COL11A1基因突變引起,主要伴有胎兒軟骨發育不良而無眼部異常[7],亦有人認為該型是Stickler綜合征的嬰兒型。基因檢測能確診該病,并能明確其分型以幫助治療[8]。但遺憾本例患兒家長拒絕行基因檢測。在缺少基因檢測的情況下該病與其他多種疾病的鑒別診斷尤為重要。