引用本文: 張桐梅, 韓梅, 應銘, 郝朋, 韓瑞芳. 家族性滲出性玻璃體視網膜病變患者基因突變檢測結果及臨床特征分析. 中華眼底病雜志, 2018, 34(6): 556-561. doi: 10.3760/cma.j.issn.1005-1015.2018.06.007 復制
家族性滲出性玻璃體視網膜病變(FEVR)的遺傳方式有常染色體顯性遺傳、常染色體隱性遺傳和伴X連鎖遺傳[1, 2]。目前發現與FEVR相關的基因分別是Norrie病(NDP)、卷曲蛋白4(FZD4)、低密度脂蛋白受體相關蛋白5(LRP5)、四旋蛋白12(TSPAN12)、驅動蛋白家族成員11(KIF11)、鋅指蛋白408(ZNF408)基因[1-4]。其中,NDP、FZD4、LRP5、TSPAN12等4個基因編碼的蛋白共同參與Wnt信號通路。該通路在控制視網膜血管生成和發育過程中發揮著重要的作用;一旦發生突變造成損傷會引起血管生長缺陷,進而引起不同表型的FEVR和類似遺傳性疾病[4]。而KIF11和ZNF408基因則參與Norrin/β-連環蛋白信號通路[2]。本研究對4個FEVR家系的NDP、FZD4、LRP5、TSPAN12基因序列進行分析,現將結果報道如下。
1 對象和方法
回顧性系列病例研究。本研究獲天津市眼科醫院倫理委員會批準;嚴格遵守赫爾辛基宣言,所有受試者或未成年監護人均簽署知情同意書。
2012年1月至2015年12月在天津市眼科醫院就診的FEVR患者9例18只眼及家系正常成員5名納入研究。患者分別來自4個無血緣關系家系。患者中,男性5例10只眼,女性4例8只眼;均為雙眼。年齡5~47歲,平均年齡(18.89±15.87)歲。2例為同卵雙胞胎。家系正常成員中,男性1名,女性4名;年齡22~46歲。
詳細收集患者及家系成員的基本信息及現病史,繪制家系圖;孟德爾遺傳規律判斷遺傳方式。所有受檢者均行最佳矯正視力(BCVA)、裂隙燈顯微鏡、眼底彩色照相、熒光素眼底血管造影(FFA)檢查。5名家系正常成員眼部檢查均正常。參照文獻[5]標準確立FEVR診斷及分期。
抽取受試者外周靜脈血3~5 ml,乙二胺四乙酸抗凝,按照羅氏DNA分離試劑盒抽提DNA提取全基因組DNA,?80 ℃冰箱保存。所有基因序列參考美國國立生物技術信息中心(NCBI)(http://www.ncbi.nlm.nih.gov/pubmed/)網站。FZD4: NG_011752.1 for gDNA, NM_012193.3 for mRNA, and NP_036325.2 for protein; LRP5: NG_015835.1 for gDNA, NM_002335.2 for mRNA, and NP_002326.2 for protein;NDP: NG_009832.1 for gDNA, NM_000266.3 for mRNA, and NP_000257.1 for protein;TSPAN12: NG_023203.1 for gDNA, NM_012338.3 for mRNA, and NP_036470.1 for protein。應用Primer 5.0軟件,設計針對候選致病基因NDP、FZD4、LRP5、TSPAN12基因所有外顯子編碼區以及連接非轉錄區的序列引物(表1),北京賽百盛基因技術有限公司合成。聚合酶鏈反應(PCR)條件:95 ℃預變性3 min,95 ℃變性30 s,63~53 ℃退火30 s,72 ℃延伸45 s,共20個循環;延伸7 min。應用膠回收試劑盒對PCR擴增產物進行純化。
應用BigDye Terminator ver.1.1 (美國Applied Biosystems公司)進行序列分析。采用美國Applied Biosystems公司3100 Genetic測序儀對PCR擴增產物進行正、反雙向DNA測序。采用Blast軟件(available at http://www.ncbi.nlm.nih.gov/blast)對測序結果和GenBank數據庫里的NDP、FZD4、LRP5、TSPAN12基因序列進行對比,查找出單核苷酸純和變異以及插入和缺失變異,得到候選基因的全部變異。對于檢測出的單核苷酸變異,查閱人類基因突變數據庫(HGMD)進行比對,初步確定DNA突變位點。在線工具PolyPhen-2(http://genetics.bwh.harvard.edu/pph2/)、SIFT(http://sift.jcvi.org/www/SIFT_enst_submit.html)預測致病性。以50名無血緣關系的健康個體為對照組。
2 結果
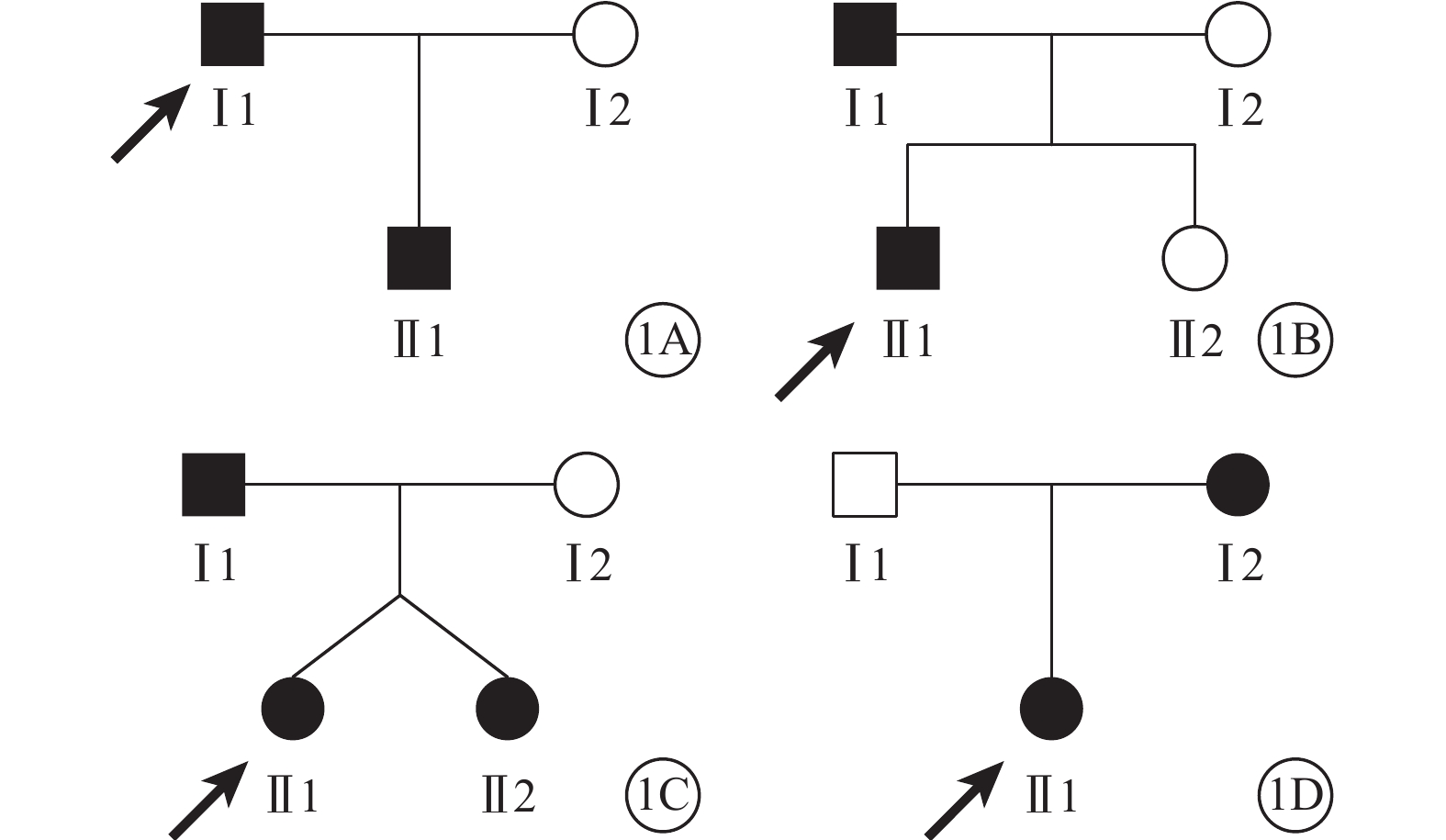
4個家系中,家系1、2為常染色體顯性遺傳,家系3、4可能為常染色體顯性遺傳或伴X連鎖遺傳(圖1)。
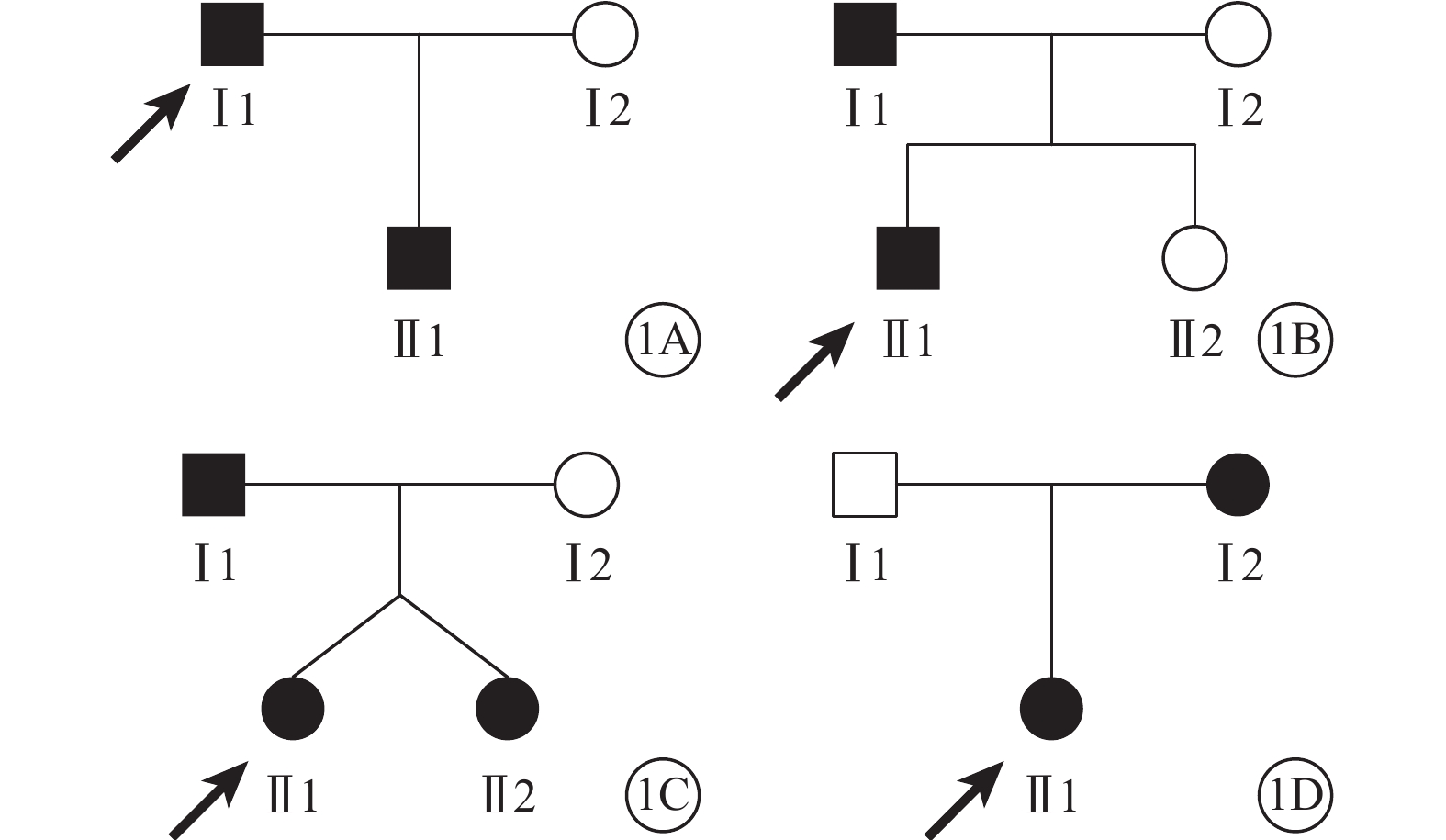 圖1
9例FEVR患者家系圖。1A~1D.家系1~4。■:男性患者;●:女性患者;↗:先證者;□:正常男性;○:正常女性。家系3Ⅱ1和Ⅱ2為同卵雙胞胎
圖1
9例FEVR患者家系圖。1A~1D.家系1~4。■:男性患者;●:女性患者;↗:先證者;□:正常男性;○:正常女性。家系3Ⅱ1和Ⅱ2為同卵雙胞胎
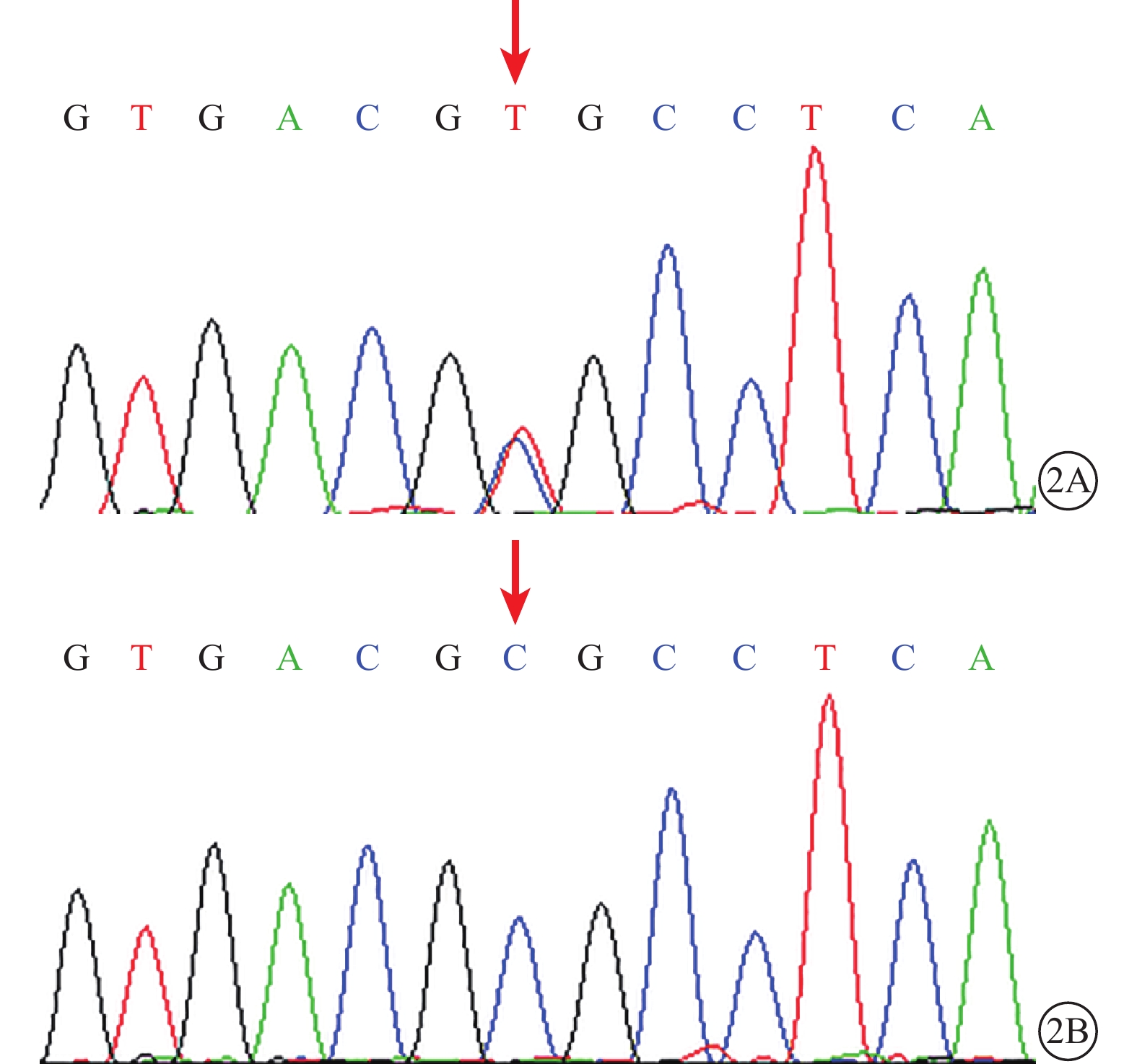
DNA測序結果發現,家系2(Ⅰ1、Ⅱ1)、3(Ⅰ1、Ⅱ1、Ⅱ2)、4(Ⅰ2、Ⅱ1)患者LRP5基因第2、18外顯子存在錯義突變,分別為c.266A>G(p.Q89R)、c.3989C>T(p.A1330V);均為單核苷酸多態性(SNP),具有普遍性。家系2(Ⅰ1、Ⅱ1)患者第6外顯子存在c.1330C>T(p.R444C)錯義突變;c.1330C>T所對應的氨基酸改變為LRP5基因所編碼蛋白的第444號氨基酸從精氨酸突變為半胱氨酸(p.R444C)(圖2A)。Polyphen2程序預測分析結果顯示,蛋白替換分值為0.882;SIFT分析提示,c.1330C>T(p.R444C)為致病性突變(表2)。家系1、3、4及50名正常對照組受檢者未檢出該基因突變(圖2B)。所有家系受檢者FZD4、NDP、TSPAN12基因均未檢測到錯義突變。
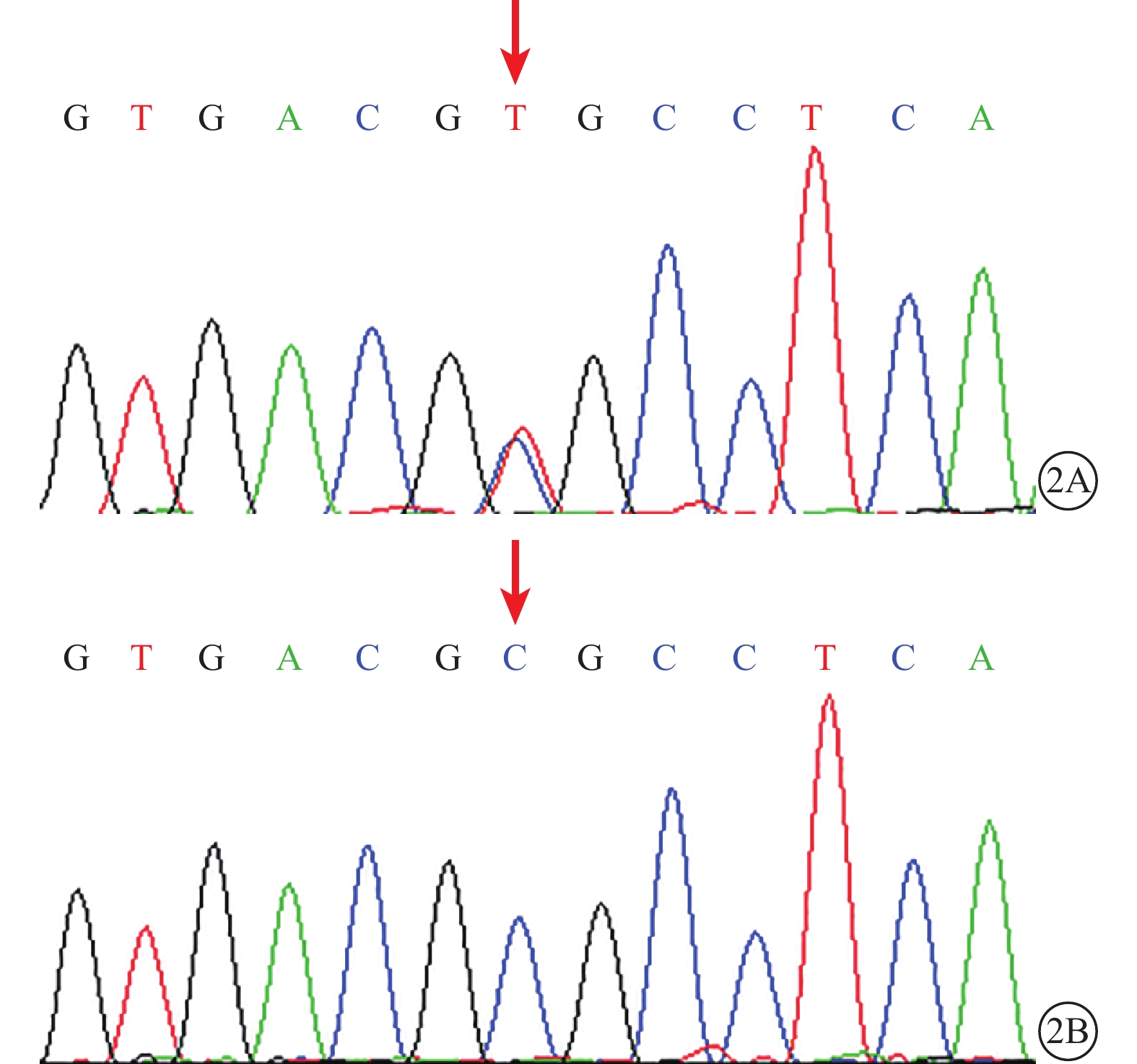 圖2
LRP5基因測序圖。2A.家系2先證者(Ⅱ1),LRP5基因第6外顯子的c.1330C>T(p.R444C)錯義突變(紅箭);2B. 對照組受檢者,該位點未見突變
圖2
LRP5基因測序圖。2A.家系2先證者(Ⅱ1),LRP5基因第6外顯子的c.1330C>T(p.R444C)錯義突變(紅箭);2B. 對照組受檢者,該位點未見突變
家系中患者LRP5基因發現同義突變5個,其中第20號外顯子c.4311C>T(p.F1437F)為新發現同義突變(表3)。所有同義突變均為SNP,具有普遍性。
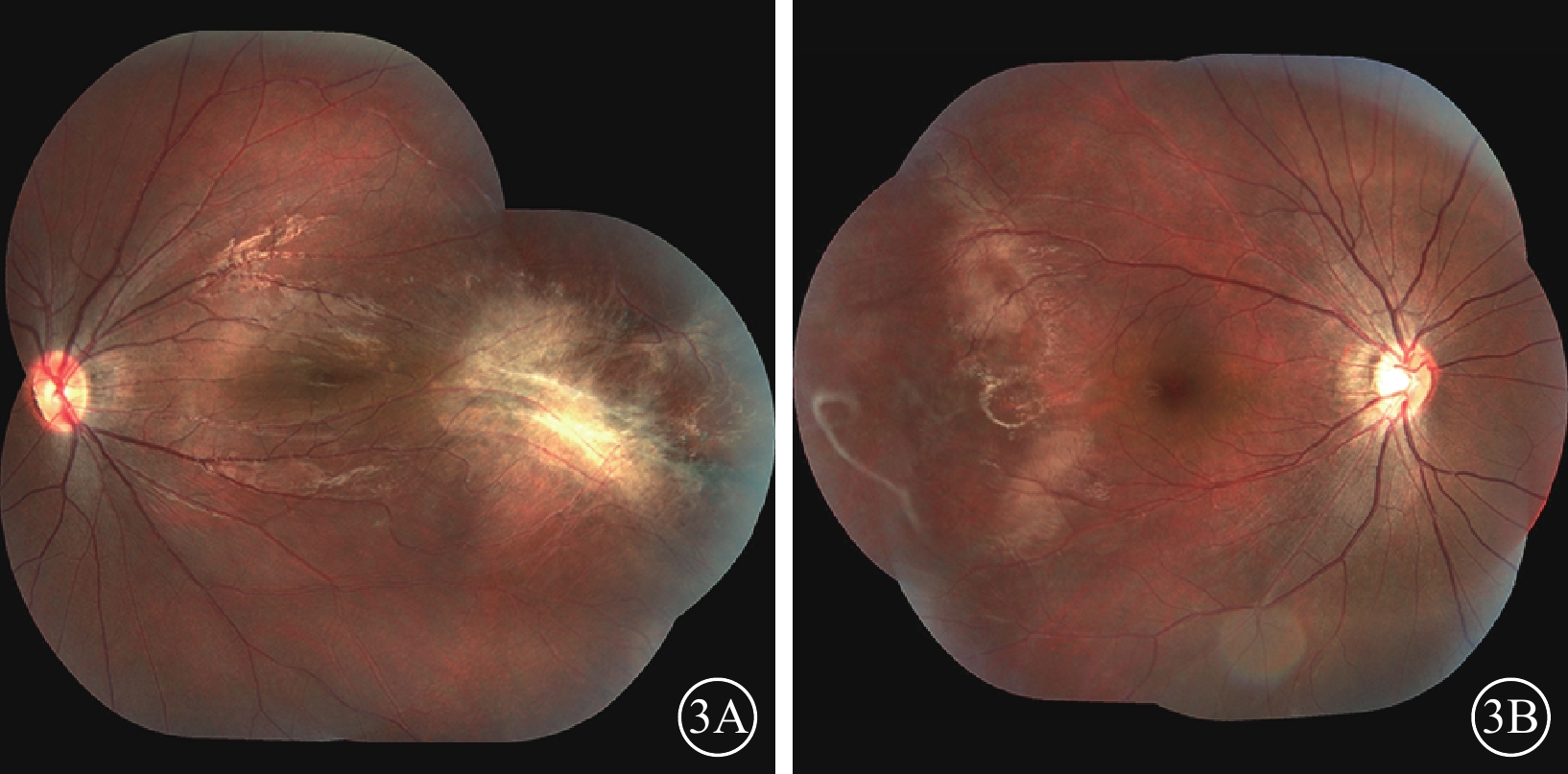
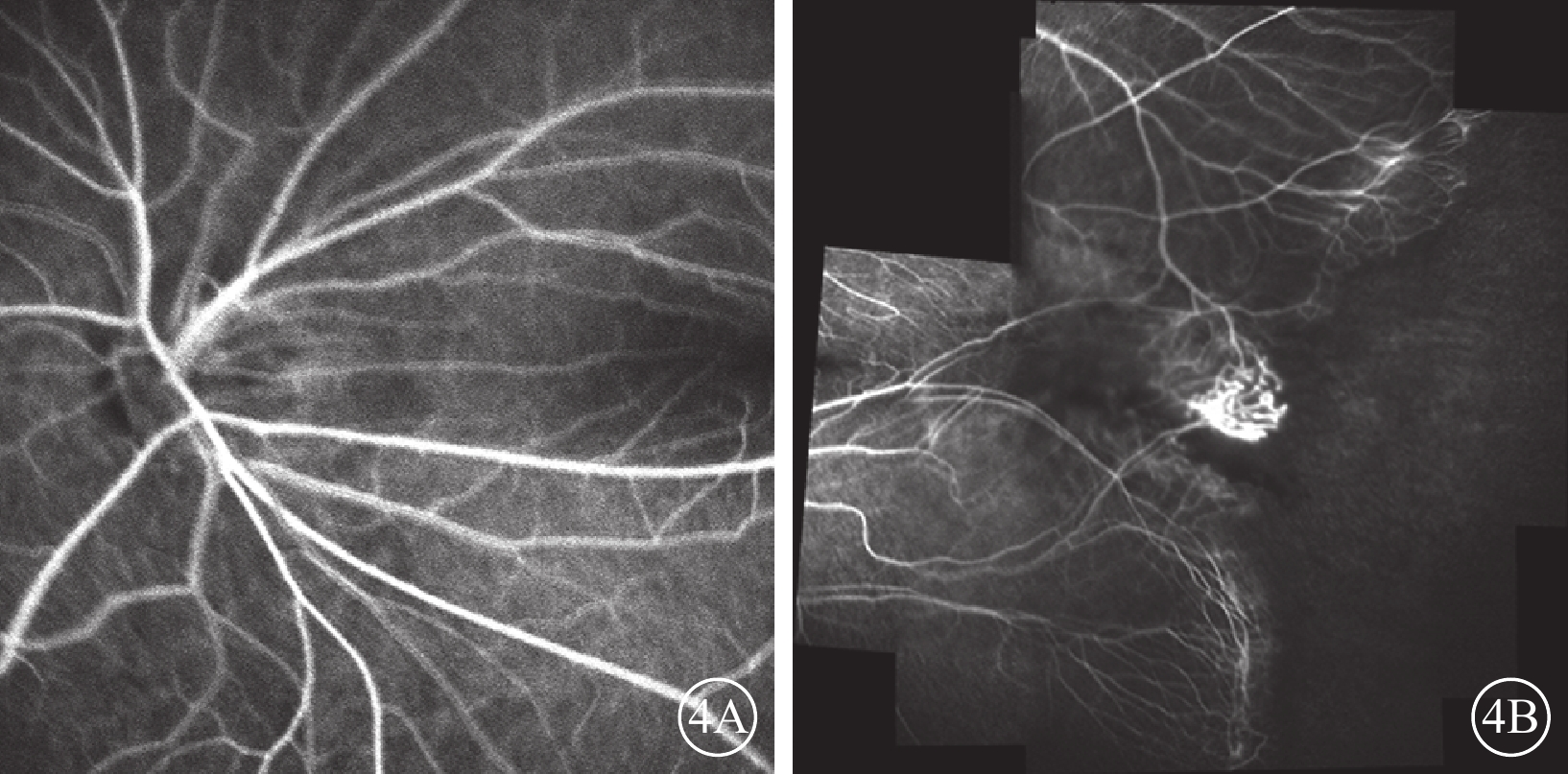
家系2先證者(Ⅱ1),男,7歲。足月順產,否認吸氧史。3歲時因視力差配戴眼鏡并行雙眼弱視訓練,效果不佳。4歲時,右眼BCVA 0.3,等效球鏡度數?8.63 D;左眼BCVA 0.5,等效球鏡度數?2.00 D。右眼后極部無明顯異常,顳側赤道部少許色素沉著;左眼視盤邊界清楚,顏色尚可;黃斑區可見機化物,少許色素沉著,視網膜血管牽拉移位。光相干斷層掃描檢查,黃斑水腫。5歲時,右眼BCVA 0.25,等效球鏡度數?9.38 D;左眼BCVA 0.4,等效球鏡度數?2.25 D。7歲時,右眼BCVA 0.3,等效球鏡度數?7.75 D;左眼BCVA 0.7,等效球鏡度數?4.88 D。右眼顳側視網膜血管走形平直,周邊血管發育停滯,末端血管紆曲,呈樹枝狀,纖維血管形成;左眼視網膜血管紆曲,黃斑區顳側纖維血管形成瘢痕,色素增生,黃斑向顳側牽拉(圖3A,3B)。FFA檢查,左眼視盤、黃斑向顳側牽拉,顳側視網膜血管走行平直,呈毛刷狀,并見動靜脈短路,周邊可見新生血管及大片無灌注區(圖4A,4B)。其父親(Ⅰ1)雙眼BCVA 1.0。眼前節未見異常。后極部未見明顯異常,顳側周邊視網膜血管走行較直。FFA檢查,雙眼視網膜周邊血管異常,走行平直,呈毛刷狀。
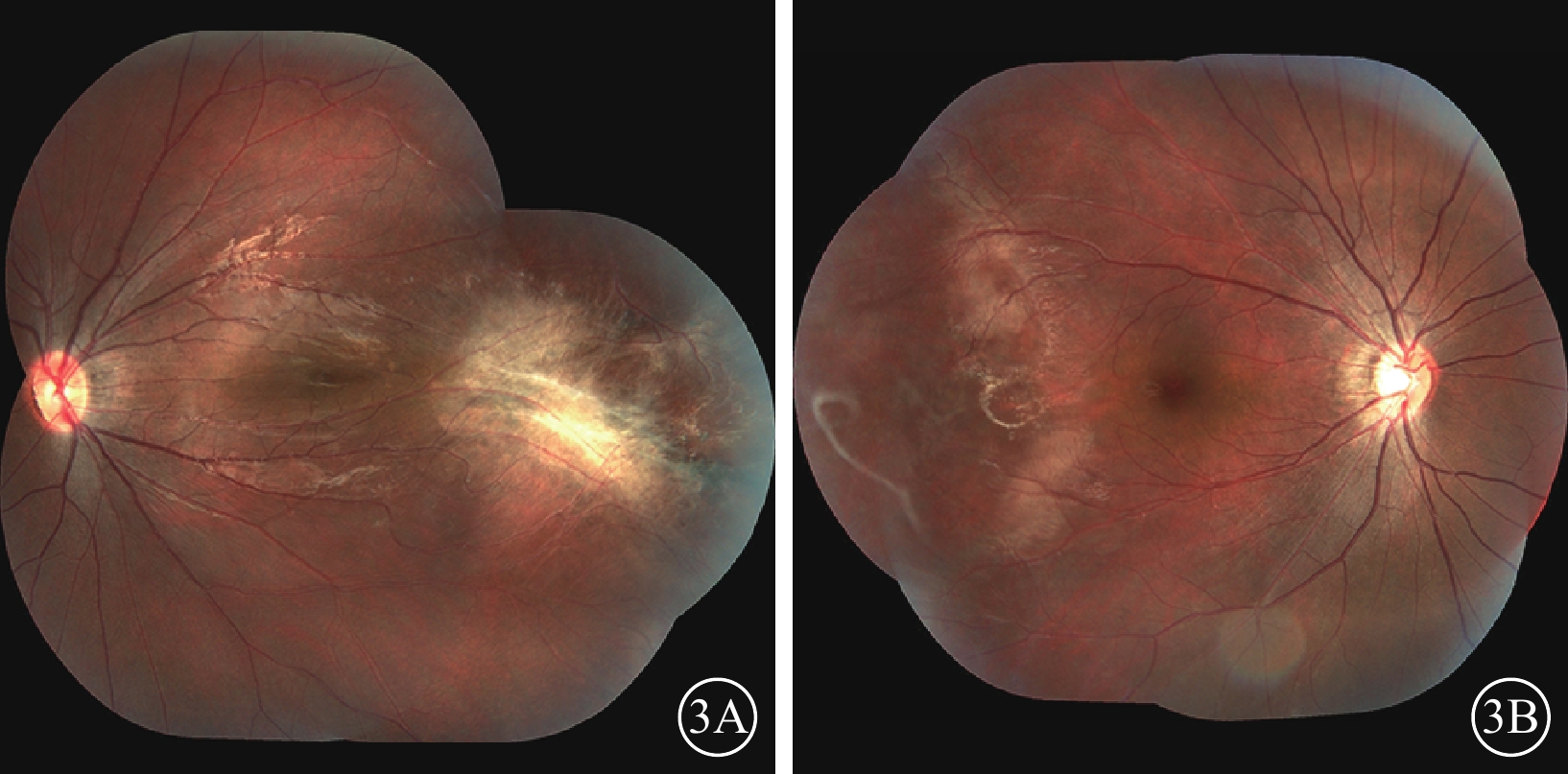 圖3
家系2先證者(Ⅱ1)雙眼彩色眼底像。3A.左眼,視網膜血管紆曲,黃斑區顳側纖維血管形成瘢痕,色素增生,黃斑向顳側牽拉;3B.右眼,顳側血管走形平直,周邊血管發育停滯,末端血管紆曲,呈樹枝狀,纖維血管形成
圖3
家系2先證者(Ⅱ1)雙眼彩色眼底像。3A.左眼,視網膜血管紆曲,黃斑區顳側纖維血管形成瘢痕,色素增生,黃斑向顳側牽拉;3B.右眼,顳側血管走形平直,周邊血管發育停滯,末端血管紆曲,呈樹枝狀,纖維血管形成
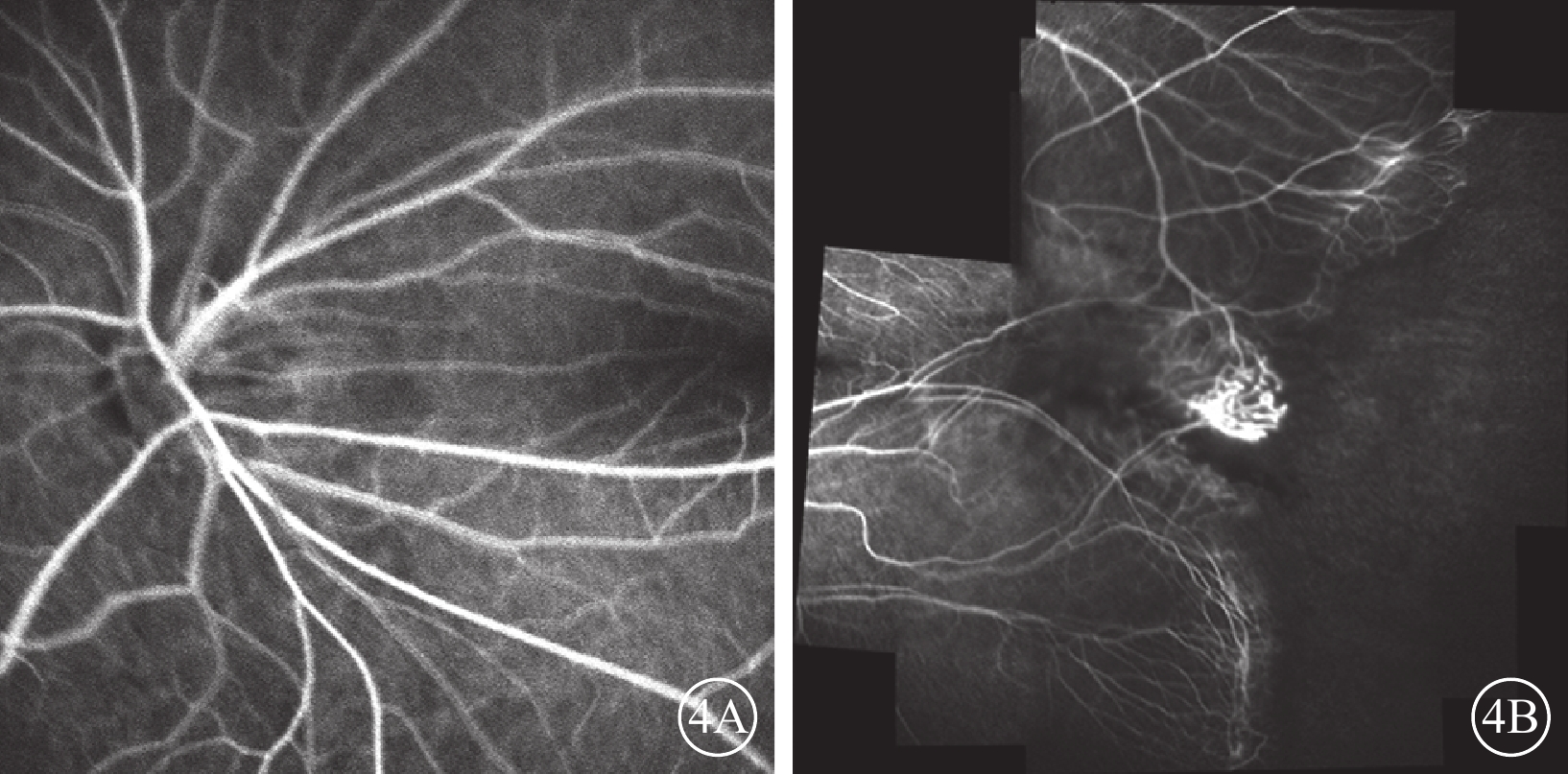 圖4
圖3A同眼FFA像。4A.視盤、黃斑向顳側牽拉;4B.顳側血管走行平直,呈毛刷狀,并見動靜脈短路,周邊可見新生血管及大片無灌注區
圖4
圖3A同眼FFA像。4A.視盤、黃斑向顳側牽拉;4B.顳側血管走行平直,呈毛刷狀,并見動靜脈短路,周邊可見新生血管及大片無灌注區
家系1先證者(Ⅰ1),男,30歲。足月產。右眼BCVA0.8,玻璃體混濁,眼底后極部未見明顯異常,顳側視網膜血管走行較直,呈毛刷狀,左眼BCVA1.0,眼底未見明顯異常。其子(Ⅱ1)7歲,足月產。右眼BCVA 0.8,等效球鏡度數?1.50 D,眼底未見明顯異常。左眼BCVA 0.5,等效球鏡度數?3.00 D;眼底視盤顏色淡,顳下視網膜血管向下方牽拉。家系3先證者(Ⅱ1、Ⅱ2)為同卵雙胞胎,女,6歲。足月產。Ⅱ1右眼BCVA 0.5,等效球鏡度數?3.25 D;眼底顳側周邊視網膜血管走行較直,呈毛刷狀。左眼BCVA 0.6,等效球鏡度數?2.50 D,眼底未見明顯異常。Ⅱ2雙眼BCVA 0.5;等效球鏡度數,右眼?3.00D,左眼?2.00 D;眼底改變同Ⅱ1。其父親(Ⅰ1),31歲。右眼眼底周邊視網膜血管走行略直,左眼未見明顯異常。家系4先證者(Ⅱ1),女,5歲。足月產。雙眼眼球震顫。右眼BCVA 0.05,等效球鏡度數?14.00 D,晶狀體后囊下混濁;眼底視盤邊界清晰、顏色尚可,黃斑區向下移位,顳下方視網膜血管變直,向下方移位。左眼BCVA 0.3,等效球鏡度數?3.00 D;視盤邊界清晰,顳側視網膜血管變直。其母親(Ⅰ2)右眼BCVA 0.5;眼底周邊視網膜血管走行略直。左眼BCVA0.3,晶狀體混濁;FFA檢查可見視網膜折疊,周邊纖維血管團,視網膜脫離。
3 討論
目前發現NDP、FZD4、LRP5、TSPAN12、KIF11以及ZNF408基因突變可導致FEVR的發生。LRP5基因位于染色體11q13.4,編碼LRP5/6。LRP5的突變可能引起常染色體顯性或隱性遺傳,而前者為FEVR患者的主要遺傳方式。目前FEVR患者中發現LRP5基因突變率為12%~18%[6]。共發現3種不同類型的LRP5基因突變:核酸缺失,包括6個不同區域的基因缺失,對蛋白結構造成不同程度影響;錯義突變,目前共發現30個不同位置的基因突變,造成編碼蛋白的改變[6-8];剪切位點改變,包括堿基插入(c.4488+2T>G)和堿基缺失(c.4489-1G>A),造成剪切功能的喪失[8]。
本研究結果顯示,家系2先證者及父親的LRP5基因6號外顯子c.1330C>T(p.R444C)發生錯義突變;FZD4、NDP以及TSPAN12均未發現基因異常突變。先證者本次就診時7歲,眼底后極部纖維血管膜形成,對黃斑形成顳側牽拉,屬2期病變。患兒3歲開始就診,隨年齡增長雙眼近視屈光度逐漸增加,考慮與兒童視覺發育相關。既往研究已證實,發育過程中視覺輸入的扭曲或不清晰可以導致近視發生[8]。其父親FFA檢查顯示雙眼視網膜周邊血管異常,其他未見明顯異常。父子臨床表型不同,其原因可能與血管發育狀態繼發改變相關。Qin等[8]報道1例20歲FEVR患者同時攜帶LRP5基因突變c.1330C>T(p.R444C)和FZD4基因突變c.1250G>A(p.R417Q)。此2個突變位點在患者家系中呈共分離現象,提示2個突變位于同一條染色體上。患者右眼鐮刀狀視網膜折疊,周邊纖維血管團;左眼后極部粘連,視網膜脫離。與此比較,本研究家系2先證者眼底病變更輕。但由于患兒年齡尚小,不排除病情的進一步進展。
Ping等[9]在2個有血緣關系的FEVR患者中發現LRP5基因上2個新的雜合突變p.A422T和p.L540P。患者和其父母均表現出典型FEVR眼底改變和輕度減少的骨密度(BMD)。其中p.A422T突變也位于6號外顯子和另一已知6號外顯子的錯義突變(c.1321G>A,p.E441K)[10]。李憶安等[11]報道的2例FEVR患者,均為FZD4與LRP5基因的聯合突變,一例為FZD4的c.283C>G聯合LRP5的c.4084A>G雜合子改變,其眼底表現為右眼視網膜皺襞連接至晶狀體后伴增生,左眼視盤血管向顳側牽引伴纖維增生;另一例為FZD4的c.1482G>A聯合LRP5的c.3538G>A雜合子改變,右眼視盤發出的鐮狀皺襞連接至晶狀體后,左眼視網膜脫離。攜帶兩個LRP5基因突變的患者與其他患者相比臨床表現更為嚴重,然而并沒有報道同時攜帶FZD4和LRP5基因的突變會引起更嚴重的表型[12]。本研究家系4的2例患者均表現為較嚴重的晶狀體混濁和眼底表現,但未發現候選基因的突變,考慮可能為其他未篩查的基因存在突變,不排除復合基因突變的可能。
FEVR發病相關的其他基因:(1)NDP基因編碼Norrin蛋白,該基因位于人類染色體Xp11.4[13]。其與Norrie病、FEVR、ROP以及原始永存玻璃體增生癥相關,FEVR患者主要表現為X染色體隱性遺傳。(2)FZD4基因編碼胞膜受體Fz卷曲蛋白家族,該基因位于染色體11q14.2,FEVR患者主要表現為常染色體顯性遺傳[14]。(3)TSPAN12基因位于常染色體7q31[15],編碼的四次跨膜蛋白與Norrin或LRP5相互作用[16]。TSPAN12位點的突變可導致常染色體顯性遺傳的FEVR[17]。(4)KIF11基因位于常染色體10q23.33。目前發現KIF11基因的突變引起的FEVR遺傳方式為常染色體顯性遺傳[1]。(5)ZNF408位于染色體11p11.2。基因突變導致的FEVR表現為常染色體顯性遺傳[2]。
雖然本研究突變基因經Polyphen2和SIFT評估為致病性,但該致病基因對氨基酸以及蛋白質的具體影響尚需進一步研究證實。4個家系中僅有1個家系篩查出突變基因,是否存在未篩查的基因與LRP5共同致病,仍需探討。本次研究的候選基因重點為Wnt信號通路的成員,其他家系也未發現突變位點存在,擴大篩查基因的范圍也是更進一步研究的方向。
家族性滲出性玻璃體視網膜病變(FEVR)的遺傳方式有常染色體顯性遺傳、常染色體隱性遺傳和伴X連鎖遺傳[1, 2]。目前發現與FEVR相關的基因分別是Norrie病(NDP)、卷曲蛋白4(FZD4)、低密度脂蛋白受體相關蛋白5(LRP5)、四旋蛋白12(TSPAN12)、驅動蛋白家族成員11(KIF11)、鋅指蛋白408(ZNF408)基因[1-4]。其中,NDP、FZD4、LRP5、TSPAN12等4個基因編碼的蛋白共同參與Wnt信號通路。該通路在控制視網膜血管生成和發育過程中發揮著重要的作用;一旦發生突變造成損傷會引起血管生長缺陷,進而引起不同表型的FEVR和類似遺傳性疾病[4]。而KIF11和ZNF408基因則參與Norrin/β-連環蛋白信號通路[2]。本研究對4個FEVR家系的NDP、FZD4、LRP5、TSPAN12基因序列進行分析,現將結果報道如下。
1 對象和方法
回顧性系列病例研究。本研究獲天津市眼科醫院倫理委員會批準;嚴格遵守赫爾辛基宣言,所有受試者或未成年監護人均簽署知情同意書。
2012年1月至2015年12月在天津市眼科醫院就診的FEVR患者9例18只眼及家系正常成員5名納入研究。患者分別來自4個無血緣關系家系。患者中,男性5例10只眼,女性4例8只眼;均為雙眼。年齡5~47歲,平均年齡(18.89±15.87)歲。2例為同卵雙胞胎。家系正常成員中,男性1名,女性4名;年齡22~46歲。
詳細收集患者及家系成員的基本信息及現病史,繪制家系圖;孟德爾遺傳規律判斷遺傳方式。所有受檢者均行最佳矯正視力(BCVA)、裂隙燈顯微鏡、眼底彩色照相、熒光素眼底血管造影(FFA)檢查。5名家系正常成員眼部檢查均正常。參照文獻[5]標準確立FEVR診斷及分期。
抽取受試者外周靜脈血3~5 ml,乙二胺四乙酸抗凝,按照羅氏DNA分離試劑盒抽提DNA提取全基因組DNA,?80 ℃冰箱保存。所有基因序列參考美國國立生物技術信息中心(NCBI)(http://www.ncbi.nlm.nih.gov/pubmed/)網站。FZD4: NG_011752.1 for gDNA, NM_012193.3 for mRNA, and NP_036325.2 for protein; LRP5: NG_015835.1 for gDNA, NM_002335.2 for mRNA, and NP_002326.2 for protein;NDP: NG_009832.1 for gDNA, NM_000266.3 for mRNA, and NP_000257.1 for protein;TSPAN12: NG_023203.1 for gDNA, NM_012338.3 for mRNA, and NP_036470.1 for protein。應用Primer 5.0軟件,設計針對候選致病基因NDP、FZD4、LRP5、TSPAN12基因所有外顯子編碼區以及連接非轉錄區的序列引物(表1),北京賽百盛基因技術有限公司合成。聚合酶鏈反應(PCR)條件:95 ℃預變性3 min,95 ℃變性30 s,63~53 ℃退火30 s,72 ℃延伸45 s,共20個循環;延伸7 min。應用膠回收試劑盒對PCR擴增產物進行純化。
應用BigDye Terminator ver.1.1 (美國Applied Biosystems公司)進行序列分析。采用美國Applied Biosystems公司3100 Genetic測序儀對PCR擴增產物進行正、反雙向DNA測序。采用Blast軟件(available at http://www.ncbi.nlm.nih.gov/blast)對測序結果和GenBank數據庫里的NDP、FZD4、LRP5、TSPAN12基因序列進行對比,查找出單核苷酸純和變異以及插入和缺失變異,得到候選基因的全部變異。對于檢測出的單核苷酸變異,查閱人類基因突變數據庫(HGMD)進行比對,初步確定DNA突變位點。在線工具PolyPhen-2(http://genetics.bwh.harvard.edu/pph2/)、SIFT(http://sift.jcvi.org/www/SIFT_enst_submit.html)預測致病性。以50名無血緣關系的健康個體為對照組。
2 結果
4個家系中,家系1、2為常染色體顯性遺傳,家系3、4可能為常染色體顯性遺傳或伴X連鎖遺傳(圖1)。
圖1
9例FEVR患者家系圖。1A~1D.家系1~4。■:男性患者;●:女性患者;↗:先證者;□:正常男性;○:正常女性。家系3Ⅱ1和Ⅱ2為同卵雙胞胎
DNA測序結果發現,家系2(Ⅰ1、Ⅱ1)、3(Ⅰ1、Ⅱ1、Ⅱ2)、4(Ⅰ2、Ⅱ1)患者LRP5基因第2、18外顯子存在錯義突變,分別為c.266A>G(p.Q89R)、c.3989C>T(p.A1330V);均為單核苷酸多態性(SNP),具有普遍性。家系2(Ⅰ1、Ⅱ1)患者第6外顯子存在c.1330C>T(p.R444C)錯義突變;c.1330C>T所對應的氨基酸改變為LRP5基因所編碼蛋白的第444號氨基酸從精氨酸突變為半胱氨酸(p.R444C)(圖2A)。Polyphen2程序預測分析結果顯示,蛋白替換分值為0.882;SIFT分析提示,c.1330C>T(p.R444C)為致病性突變(表2)。家系1、3、4及50名正常對照組受檢者未檢出該基因突變(圖2B)。所有家系受檢者FZD4、NDP、TSPAN12基因均未檢測到錯義突變。
圖2
LRP5基因測序圖。2A.家系2先證者(Ⅱ1),LRP5基因第6外顯子的c.1330C>T(p.R444C)錯義突變(紅箭);2B. 對照組受檢者,該位點未見突變
家系中患者LRP5基因發現同義突變5個,其中第20號外顯子c.4311C>T(p.F1437F)為新發現同義突變(表3)。所有同義突變均為SNP,具有普遍性。
家系2先證者(Ⅱ1),男,7歲。足月順產,否認吸氧史。3歲時因視力差配戴眼鏡并行雙眼弱視訓練,效果不佳。4歲時,右眼BCVA 0.3,等效球鏡度數?8.63 D;左眼BCVA 0.5,等效球鏡度數?2.00 D。右眼后極部無明顯異常,顳側赤道部少許色素沉著;左眼視盤邊界清楚,顏色尚可;黃斑區可見機化物,少許色素沉著,視網膜血管牽拉移位。光相干斷層掃描檢查,黃斑水腫。5歲時,右眼BCVA 0.25,等效球鏡度數?9.38 D;左眼BCVA 0.4,等效球鏡度數?2.25 D。7歲時,右眼BCVA 0.3,等效球鏡度數?7.75 D;左眼BCVA 0.7,等效球鏡度數?4.88 D。右眼顳側視網膜血管走形平直,周邊血管發育停滯,末端血管紆曲,呈樹枝狀,纖維血管形成;左眼視網膜血管紆曲,黃斑區顳側纖維血管形成瘢痕,色素增生,黃斑向顳側牽拉(圖3A,3B)。FFA檢查,左眼視盤、黃斑向顳側牽拉,顳側視網膜血管走行平直,呈毛刷狀,并見動靜脈短路,周邊可見新生血管及大片無灌注區(圖4A,4B)。其父親(Ⅰ1)雙眼BCVA 1.0。眼前節未見異常。后極部未見明顯異常,顳側周邊視網膜血管走行較直。FFA檢查,雙眼視網膜周邊血管異常,走行平直,呈毛刷狀。
圖3
家系2先證者(Ⅱ1)雙眼彩色眼底像。3A.左眼,視網膜血管紆曲,黃斑區顳側纖維血管形成瘢痕,色素增生,黃斑向顳側牽拉;3B.右眼,顳側血管走形平直,周邊血管發育停滯,末端血管紆曲,呈樹枝狀,纖維血管形成
圖4
圖3A同眼FFA像。4A.視盤、黃斑向顳側牽拉;4B.顳側血管走行平直,呈毛刷狀,并見動靜脈短路,周邊可見新生血管及大片無灌注區
家系1先證者(Ⅰ1),男,30歲。足月產。右眼BCVA0.8,玻璃體混濁,眼底后極部未見明顯異常,顳側視網膜血管走行較直,呈毛刷狀,左眼BCVA1.0,眼底未見明顯異常。其子(Ⅱ1)7歲,足月產。右眼BCVA 0.8,等效球鏡度數?1.50 D,眼底未見明顯異常。左眼BCVA 0.5,等效球鏡度數?3.00 D;眼底視盤顏色淡,顳下視網膜血管向下方牽拉。家系3先證者(Ⅱ1、Ⅱ2)為同卵雙胞胎,女,6歲。足月產。Ⅱ1右眼BCVA 0.5,等效球鏡度數?3.25 D;眼底顳側周邊視網膜血管走行較直,呈毛刷狀。左眼BCVA 0.6,等效球鏡度數?2.50 D,眼底未見明顯異常。Ⅱ2雙眼BCVA 0.5;等效球鏡度數,右眼?3.00D,左眼?2.00 D;眼底改變同Ⅱ1。其父親(Ⅰ1),31歲。右眼眼底周邊視網膜血管走行略直,左眼未見明顯異常。家系4先證者(Ⅱ1),女,5歲。足月產。雙眼眼球震顫。右眼BCVA 0.05,等效球鏡度數?14.00 D,晶狀體后囊下混濁;眼底視盤邊界清晰、顏色尚可,黃斑區向下移位,顳下方視網膜血管變直,向下方移位。左眼BCVA 0.3,等效球鏡度數?3.00 D;視盤邊界清晰,顳側視網膜血管變直。其母親(Ⅰ2)右眼BCVA 0.5;眼底周邊視網膜血管走行略直。左眼BCVA0.3,晶狀體混濁;FFA檢查可見視網膜折疊,周邊纖維血管團,視網膜脫離。
3 討論
目前發現NDP、FZD4、LRP5、TSPAN12、KIF11以及ZNF408基因突變可導致FEVR的發生。LRP5基因位于染色體11q13.4,編碼LRP5/6。LRP5的突變可能引起常染色體顯性或隱性遺傳,而前者為FEVR患者的主要遺傳方式。目前FEVR患者中發現LRP5基因突變率為12%~18%[6]。共發現3種不同類型的LRP5基因突變:核酸缺失,包括6個不同區域的基因缺失,對蛋白結構造成不同程度影響;錯義突變,目前共發現30個不同位置的基因突變,造成編碼蛋白的改變[6-8];剪切位點改變,包括堿基插入(c.4488+2T>G)和堿基缺失(c.4489-1G>A),造成剪切功能的喪失[8]。
本研究結果顯示,家系2先證者及父親的LRP5基因6號外顯子c.1330C>T(p.R444C)發生錯義突變;FZD4、NDP以及TSPAN12均未發現基因異常突變。先證者本次就診時7歲,眼底后極部纖維血管膜形成,對黃斑形成顳側牽拉,屬2期病變。患兒3歲開始就診,隨年齡增長雙眼近視屈光度逐漸增加,考慮與兒童視覺發育相關。既往研究已證實,發育過程中視覺輸入的扭曲或不清晰可以導致近視發生[8]。其父親FFA檢查顯示雙眼視網膜周邊血管異常,其他未見明顯異常。父子臨床表型不同,其原因可能與血管發育狀態繼發改變相關。Qin等[8]報道1例20歲FEVR患者同時攜帶LRP5基因突變c.1330C>T(p.R444C)和FZD4基因突變c.1250G>A(p.R417Q)。此2個突變位點在患者家系中呈共分離現象,提示2個突變位于同一條染色體上。患者右眼鐮刀狀視網膜折疊,周邊纖維血管團;左眼后極部粘連,視網膜脫離。與此比較,本研究家系2先證者眼底病變更輕。但由于患兒年齡尚小,不排除病情的進一步進展。
Ping等[9]在2個有血緣關系的FEVR患者中發現LRP5基因上2個新的雜合突變p.A422T和p.L540P。患者和其父母均表現出典型FEVR眼底改變和輕度減少的骨密度(BMD)。其中p.A422T突變也位于6號外顯子和另一已知6號外顯子的錯義突變(c.1321G>A,p.E441K)[10]。李憶安等[11]報道的2例FEVR患者,均為FZD4與LRP5基因的聯合突變,一例為FZD4的c.283C>G聯合LRP5的c.4084A>G雜合子改變,其眼底表現為右眼視網膜皺襞連接至晶狀體后伴增生,左眼視盤血管向顳側牽引伴纖維增生;另一例為FZD4的c.1482G>A聯合LRP5的c.3538G>A雜合子改變,右眼視盤發出的鐮狀皺襞連接至晶狀體后,左眼視網膜脫離。攜帶兩個LRP5基因突變的患者與其他患者相比臨床表現更為嚴重,然而并沒有報道同時攜帶FZD4和LRP5基因的突變會引起更嚴重的表型[12]。本研究家系4的2例患者均表現為較嚴重的晶狀體混濁和眼底表現,但未發現候選基因的突變,考慮可能為其他未篩查的基因存在突變,不排除復合基因突變的可能。
FEVR發病相關的其他基因:(1)NDP基因編碼Norrin蛋白,該基因位于人類染色體Xp11.4[13]。其與Norrie病、FEVR、ROP以及原始永存玻璃體增生癥相關,FEVR患者主要表現為X染色體隱性遺傳。(2)FZD4基因編碼胞膜受體Fz卷曲蛋白家族,該基因位于染色體11q14.2,FEVR患者主要表現為常染色體顯性遺傳[14]。(3)TSPAN12基因位于常染色體7q31[15],編碼的四次跨膜蛋白與Norrin或LRP5相互作用[16]。TSPAN12位點的突變可導致常染色體顯性遺傳的FEVR[17]。(4)KIF11基因位于常染色體10q23.33。目前發現KIF11基因的突變引起的FEVR遺傳方式為常染色體顯性遺傳[1]。(5)ZNF408位于染色體11p11.2。基因突變導致的FEVR表現為常染色體顯性遺傳[2]。
雖然本研究突變基因經Polyphen2和SIFT評估為致病性,但該致病基因對氨基酸以及蛋白質的具體影響尚需進一步研究證實。4個家系中僅有1個家系篩查出突變基因,是否存在未篩查的基因與LRP5共同致病,仍需探討。本次研究的候選基因重點為Wnt信號通路的成員,其他家系也未發現突變位點存在,擴大篩查基因的范圍也是更進一步研究的方向。