引用本文: 白周現, 焦智慧, 孔祥東. 一例腎-視神經乳頭缺損綜合征患兒基因突變檢測分析. 中華眼底病雜志, 2018, 34(6): 552-555. doi: 10.3760/cma.j.issn.1005-1015.2018.06.006 復制
腎-視神經乳頭缺損綜合征(RCS)是一種罕見的常染色體顯性遺傳病,以眼部和腎臟的不同程度異常為特征。PAX2基因突變導致的RCS眼部表型主要為不易察覺的視盤發育異常。該異常典型特征為視盤缺損以及從其外圍輻射發出明顯的視網膜血管,因形態上像牽牛花而被稱為“牽牛花綜合征”[1-2]。該病的腎臟異常主要表現為先天性腎臟和泌尿系統畸形(CAKUT)[3]。這種綜合征在有遺傳史的家族內和家族間表現出很大的臨床異質性。關于RCS國內僅有少數個案報告[4-5],尚無在基因水平確診的報道。為明確一例右眼白內障、左眼先天性視盤發育不良患兒的遺傳學致病原因并幫助確診,我們選取了眼病相關遺傳致病基因組成的二代測序基因包對該家系進行了基因測序分析,確認其為PAX2基因雜合突變所致的RCS。現將結果報道如下。
1 對象和方法
本研究獲鄭州大學第一附屬醫院倫理委員會批準;嚴格遵守赫爾辛基宣言,所有受試者及未成年受試者監護人均簽署知情同意書。
患兒男,5歲。因視力繼續下降、眼科未確診于2018年1月來本院行遺傳學診斷。患兒足月剖腹產,出生體重3.15 kg。出生后因肺炎、呼吸衰竭住院20 d,吸氧史(+)。當地醫院眼部CT檢查發現左眼球后占位。3月齡時發現右眼白瞳3 d。2013年7月因右眼先天性白內障在外院接受右眼白內障吸除、人工晶狀體(IOL)植入、前部玻璃體切割手術。手術前患兒右眼角膜透明,晶狀體全混濁,視網膜未見明顯異常;左眼眼前節正常,角膜發育不良,視盤缺損且視盤外圍輻射發出視網膜血管(圖1),周邊視網膜未見異常。手術后右眼視軸區清亮。
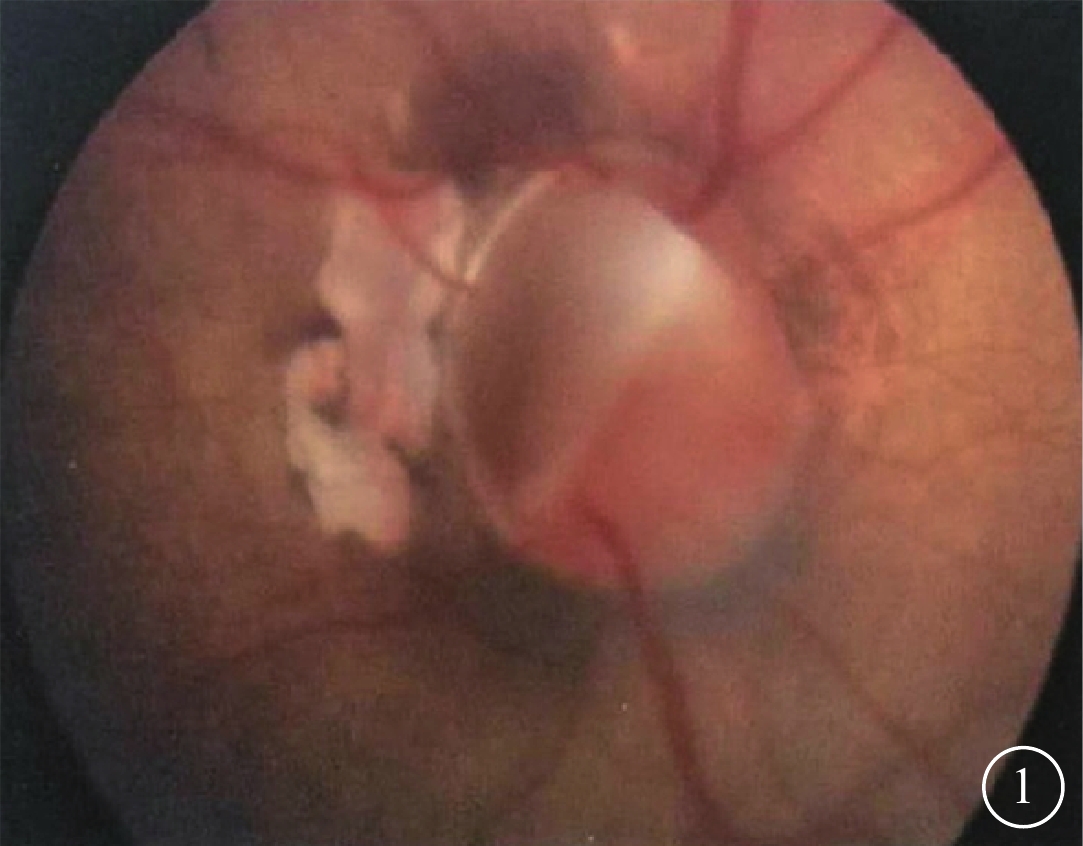
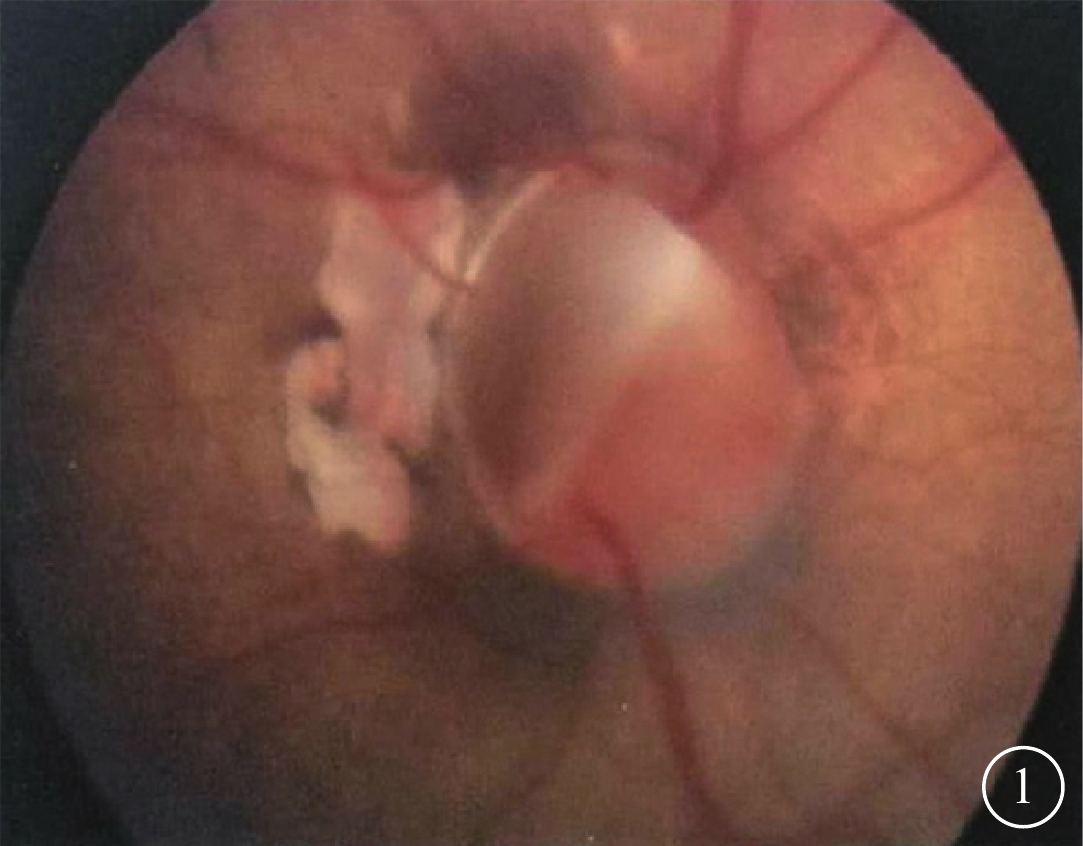 圖1
患兒左眼彩色眼底像。可見視盤缺損以及視盤外圍輻射發出的視網膜血管。
圖1
患兒左眼彩色眼底像。可見視盤缺損以及視盤外圍輻射發出的視網膜血管。
采集患兒及其正常表型的父母外周靜脈血4 ml,乙二胺四乙酸抗凝。采用磁珠法提取試劑盒(艾本德中國有限公司)和自動化DNA提取設備抽提基因組DNA。利用眼科疾病基因檢測包混合引物(含381個基因,北京邁基諾基因科技有限責任公司合成),通過本科室測序平臺Illumina NextSeq 500進行基因測序。人類基因組參考序列版本選擇GRCh37(hg19)。
對二代測序突變結果進行Sanger測序法驗證。以患兒及其父母提取的基因組DNA為模板,使用Taq DNA聚合酶(立陶宛Fermentas公司)和引物(GeneTool軟件設計)對目標序列進行擴增。正向引物5′-TCCGCCCTGCCTGCTTCCTTCG-3′,反向引物5′-CAGGGCTGGCCGGGGAAGTAGG-3′,擴增片段長度為330堿基對(bp)。擴增條件為:95 ℃預變性3 min;95 ℃變性30 s,60 ℃退火30 s,72 ℃延伸45 s,重復循環35次;72 ℃終延伸10 min,25 ℃室溫保存。將聚合酶鏈反應(PCR)擴增的目的DNA片段純化后,用dGTPBigDye? Terminator(熒光標記終止物試劑盒,變性1 min;96 ℃10 s,50 ℃ 5 s,60 ℃ 4 min;重復循環25次;4 ℃保溫。采用乙醇/醋酸鈉純化,按BigDyeV3.1操作手冊進行。將純化產物變性后上測序儀(美國ABI公司)進行序列分析。
利用人類基因變異數據庫(HGMD)等工具查詢目標突變位點是否已被收錄。通過PubMed數據庫檢索是否有相關論文報道,被學術論文報道過的相關致病變異可確定為致病性突變。查詢ClinVar數據庫(https://www.ncbi.nlm.nih.gov/clinvar/)對本研究突變的收錄情況,確定人類基因變異和表型的關聯情況和證據。檢索基因組整合數據庫(gnomAD)以明確該突變在總人群中的出現頻率。Mutationt@sting蛋白預測軟件(http://mutationtaster.org/)預測插入突變對蛋白質功能的影響,以判斷其致病性。依據《遺傳變異分類標準與指南》[6]和2015年版的《ACMG Standard and Guidelines》[7]對本研究檢出突變的致病性進行判斷。
2 結果
Illumina測序平臺DNA測序發現,患兒PAX2基因存在c.70dupG(p.V26Gfs*28)雜合突變,導致PAX2基因的編碼蛋白轉錄因子配對盒2的第26位氨基酸由纈氨酸突變為甘氨酸并移碼28個密碼子后終止翻譯。正常表型的父母均不攜帶此變異。
Sanger測序法驗證發現,患兒及其父母的PAX2基因突變情況與二代測序一致(圖2)。患兒疾病表型推斷為PAX2基因c.70dupG(p.V26Gfs*28)雜合突變所致,系常染色體顯性遺傳。
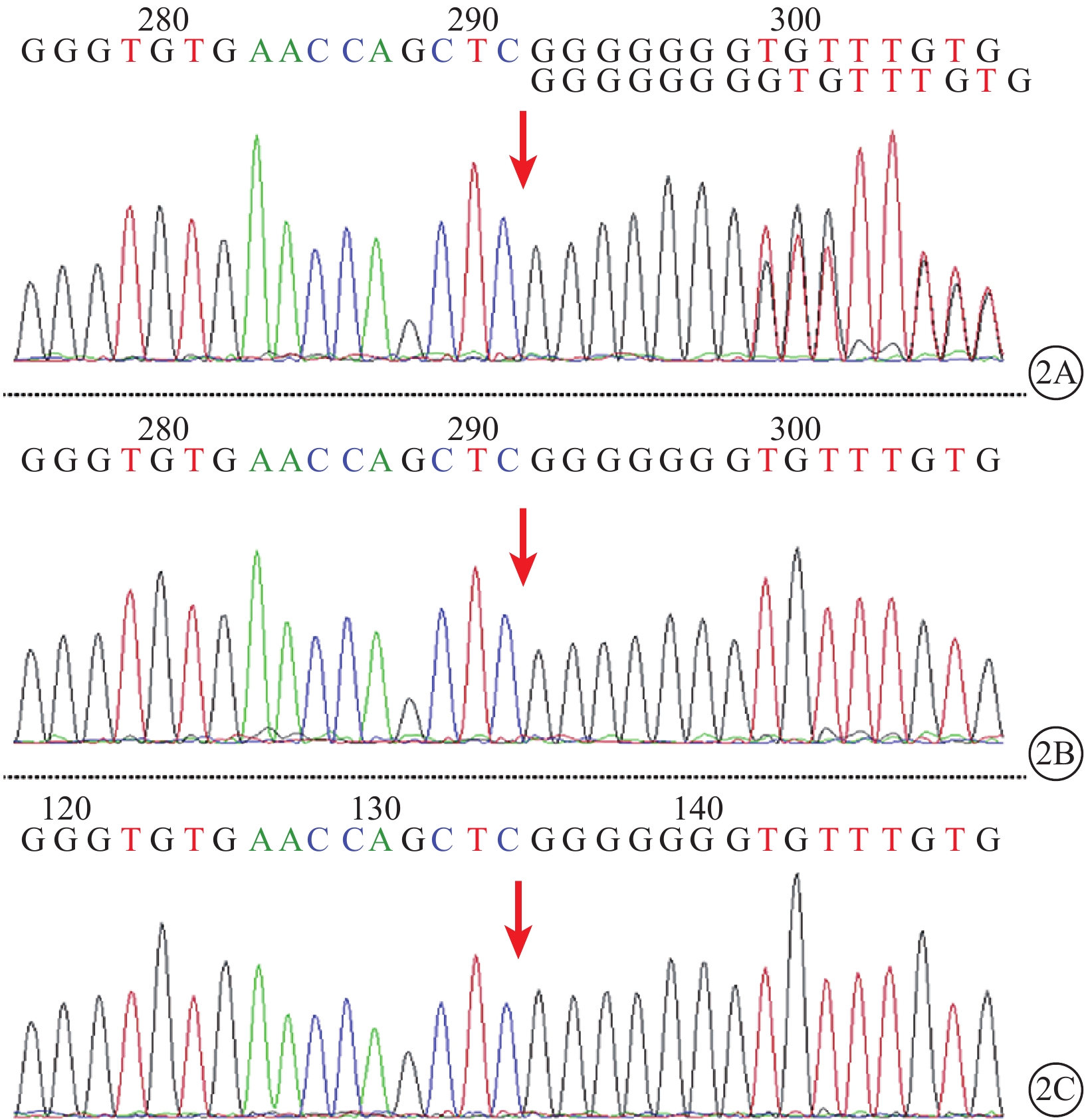
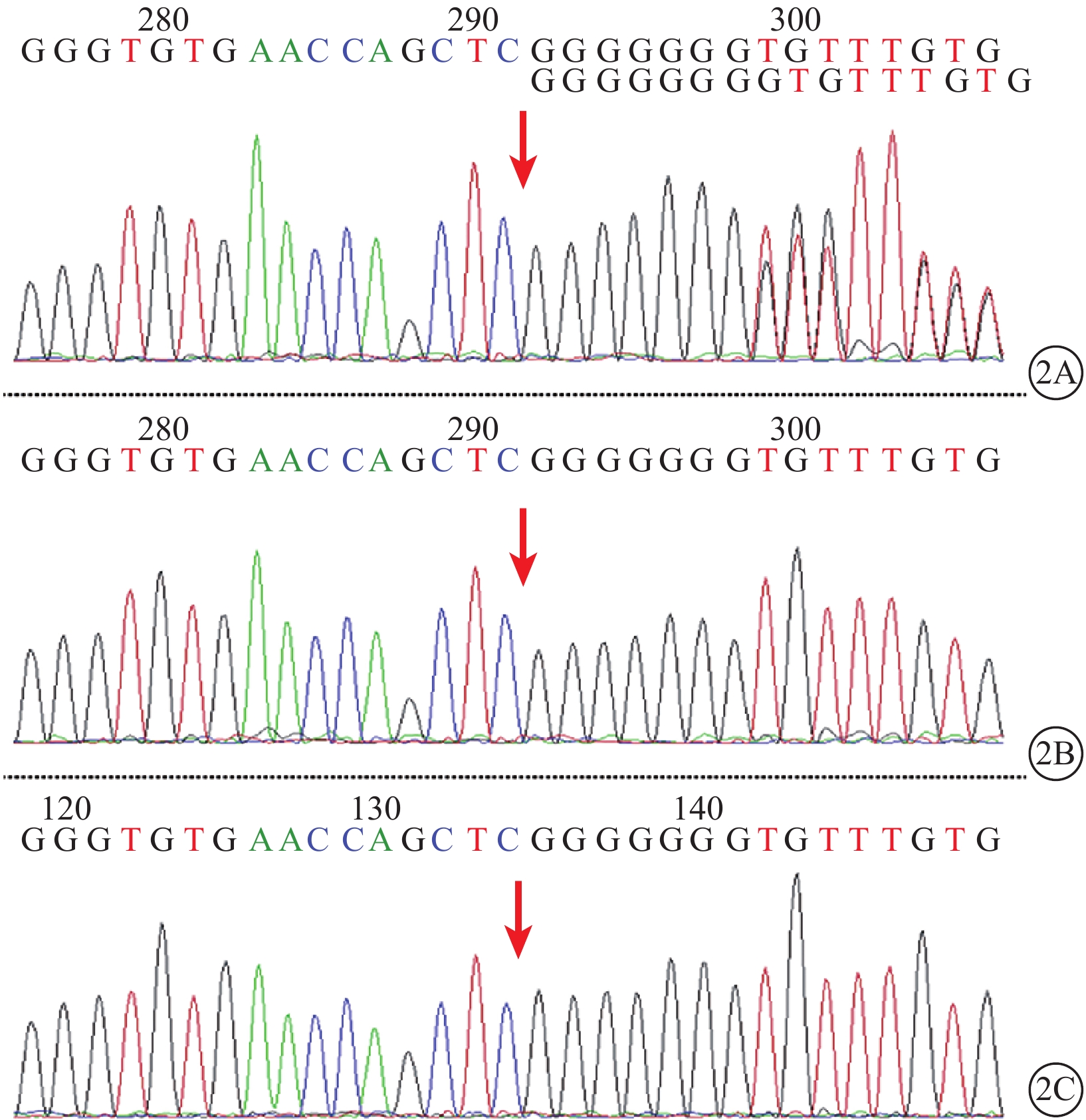 圖2
PAX2基因c.70dupG(p.V26Gfs*28)突變位點Sanger測序驗證結果。2A.患兒;2B.患兒父親;2C. 患兒母親。患兒PAX2基因存在c.70dupG(p.V26Gfs*28)雜合突變,導致PAX2基因的編碼蛋白轉錄因子配對盒2的第26位氨基酸由纈氨酸突變為甘氨酸并移碼28個密碼子后終止翻譯(紅箭)
圖2
PAX2基因c.70dupG(p.V26Gfs*28)突變位點Sanger測序驗證結果。2A.患兒;2B.患兒父親;2C. 患兒母親。患兒PAX2基因存在c.70dupG(p.V26Gfs*28)雜合突變,導致PAX2基因的編碼蛋白轉錄因子配對盒2的第26位氨基酸由纈氨酸突變為甘氨酸并移碼28個密碼子后終止翻譯(紅箭)
HGMD數據庫專業版和PubMed檢索發現PAX2基因c.70dupG(p.V26Gfs*28)突變致病性已有報道。人類基因變異和表型間關系的數據庫ClinVar亦有本研究中突變的致病性記錄,與局灶節段性腎小球硬化癥7型[OMIM:616002]、RCS[OMIM:120330]發病相關。利用Mutationt@sting蛋白質預測軟件對此位點進行預測分析,結果顯示為有害突變(disease causing),主要表現為移碼導致蛋白質翻譯提前終止而產生了截斷的無功能產物蛋白。根據《遺傳變異分類標準與指南》和2015年版《ACMG Standard and Guidelines》判斷該移碼突變為致病性變異,致病性證據非常強(等級PVS1)。gnomAD顯示該突變的dbSNP ID為rs768607170,僅在猶太人、非洲人和非芬蘭裔歐洲人中各出現過1例,人群總頻率約為十萬分之一;尚未在中國人或東亞人群中發現過。
2018年3月,患兒根據遺傳分析結果和建議在本院行針對性腎臟彩色超聲和腎功能檢查。腎臟彩色超聲檢查發現,雙腎大小形態正常;左腎實質可見大小約5.0 mm×4.8 mm囊性回聲,邊界清楚,后方回聲增強,左腎集合系統輕度分離,腎盂前后徑約8.0 mm;左腎小囊腫,雙腎彌漫性回聲改變。其余正常。腎功能檢查結果顯示,α1微球蛋白指標升高,其他各項指標在正常范圍內。提示患兒存在輕度腎臟異常或RCS綜合征腎臟病變的早期表現,需要定期復查及觀察。
3 討論
本例患兒眼部主要表現為右眼先天性白內障以及左眼視盤凹陷、發育不全、眼底“牽牛花綜合征”、視力持續下降等;腎部異常包括雙腎彌漫性回聲改變、左腎集合系統輕度分離、左腎小囊腫等特征性改變。患者這些臨床表型符合RCS的典型特征。研究者認為,RCS一般屬于PAX2基因突變引起的單基因遺傳病。根據孟德爾遺傳定律,本家系屬于常染色體顯性遺傳,針對常見的眼病遺傳病致病基因進行靶向捕獲和二代測序來篩查突變。測序分析發現患兒PAX2基因發生c.70dupG(p.V26Gfs*28)雜合突變導致RCS。
PAX2基因位于10q24.31,由13個外顯子組成,該基因的編碼蛋白轉錄因子配對盒2是轉錄因子基因家族的一員,它們的中心性特征為保守的DNA結合配對盒結構域。研究發現,PAX2基因突變可導致視覺神經組織缺損和腎臟發育不全,OMIM數據庫收錄PAX2基因為RCS[OMIM:120330]的致病基因。本研究檢出的PAX2基因c.70dupG(p.V26Gfs*28)雜合突變,在編碼鏈第70個核苷酸(第2外顯子)后插入了一個鳥苷酸(G)導致其產生了截斷的無功能蛋白產物。該患兒檢出的突變位于連續的7個鳥苷酸(G)這個突變熱點區域。Sanyanusin等[8]報道的PAX2基因c.76dupG(p.Val26Glyfs*28)突變和Thomas等[9]報道的PAX2基因c.69_70insG(G24fs*28)移碼突變與本例患兒PAX2基因c.70dupG(p.V26Gfs*28)的突變為同一突變。由于該突變所在位置連續的7個鳥苷酸(G)的特殊性,在檢索以往的研究文獻時需要注意該突變的書寫問題。
RCS有臨床表現異質性。相關眼部異常還可能包括角膜直徑小、視網膜缺損、鞏膜葡萄腫、視神經囊腫、小眼畸形和色素性黃斑發育不良等[1]。回顧PAX2基因突變和RCS的研究發現,本研究檢出突變的上下游10 bp區域內有多個已知致病性突變[10-13],推斷它可能為突變活躍區域。這些致病性突變在物理位置上相緊鄰,在蛋白結構中可能具有相近的作用。有一些PAX2基因突變導致的RCS患者伴隨CAKUT但沒有明顯的眼部異常,或不伴隨CAKUT但有眼部異常[14-15]。這種異質性不僅表現在不同家系的患者,甚至表現在相同基因突變的家系內部。Rieger[15]報道過一個RCS家系父子女3人表現出了很大的臨床表型差異。該家系中,父親表現為雙側視盤異常,最后因慢性腎炎死亡;兒子表現為黃斑和視網膜異常,但有正常的腎功能;女兒表現為正常眼,最后卻因進行性腎衰竭而死亡。本例患兒3月齡時因右眼先天性白內障而發現RCS眼部異常。經遺傳學分析,發現具有致病性的PAX2基因c.70dupG(p.V26Gfs*28)突變。根據遺傳學診斷結果,患兒經針對性的腎臟檢查發現雙腎彌漫性回聲改變、左腎集合系統輕度分離、左腎小囊腫等異常。遺傳學分析指導我們在該患兒腎臟出現臨床癥狀前發現了輕度異常或早期病變,這對于疾病的早期干預和治療具有重要意義。患兒右眼先天性白內障表型以往研究報道未見描述,這可能是PAX2基因突變導致的RCS綜合征臨床異質性的體現。
本研究從遺傳學角度分析了一例RCS患兒的致病原因,結合PAX2基因c.70dupG(p.V26Gfs*28)雜合突變的致病性和相關臨床檢查結果確認了一例RCS病例,該發現拓展了PAX2基因突變常染色體顯性遺傳RCS的表型譜,為理解該病的臨床表現和分子機制提供了有用信息。本研究PAX2基因致病突變的檢出和致病性分析表明,臨床異質性較大的遺傳性疾病的分子診斷對那些尚無明顯臨床表現的病變的早期發現及早期干預和治療具有重要意義。
腎-視神經乳頭缺損綜合征(RCS)是一種罕見的常染色體顯性遺傳病,以眼部和腎臟的不同程度異常為特征。PAX2基因突變導致的RCS眼部表型主要為不易察覺的視盤發育異常。該異常典型特征為視盤缺損以及從其外圍輻射發出明顯的視網膜血管,因形態上像牽牛花而被稱為“牽牛花綜合征”[1-2]。該病的腎臟異常主要表現為先天性腎臟和泌尿系統畸形(CAKUT)[3]。這種綜合征在有遺傳史的家族內和家族間表現出很大的臨床異質性。關于RCS國內僅有少數個案報告[4-5],尚無在基因水平確診的報道。為明確一例右眼白內障、左眼先天性視盤發育不良患兒的遺傳學致病原因并幫助確診,我們選取了眼病相關遺傳致病基因組成的二代測序基因包對該家系進行了基因測序分析,確認其為PAX2基因雜合突變所致的RCS。現將結果報道如下。
1 對象和方法
本研究獲鄭州大學第一附屬醫院倫理委員會批準;嚴格遵守赫爾辛基宣言,所有受試者及未成年受試者監護人均簽署知情同意書。
患兒男,5歲。因視力繼續下降、眼科未確診于2018年1月來本院行遺傳學診斷。患兒足月剖腹產,出生體重3.15 kg。出生后因肺炎、呼吸衰竭住院20 d,吸氧史(+)。當地醫院眼部CT檢查發現左眼球后占位。3月齡時發現右眼白瞳3 d。2013年7月因右眼先天性白內障在外院接受右眼白內障吸除、人工晶狀體(IOL)植入、前部玻璃體切割手術。手術前患兒右眼角膜透明,晶狀體全混濁,視網膜未見明顯異常;左眼眼前節正常,角膜發育不良,視盤缺損且視盤外圍輻射發出視網膜血管(圖1),周邊視網膜未見異常。手術后右眼視軸區清亮。
圖1
患兒左眼彩色眼底像。可見視盤缺損以及視盤外圍輻射發出的視網膜血管。
采集患兒及其正常表型的父母外周靜脈血4 ml,乙二胺四乙酸抗凝。采用磁珠法提取試劑盒(艾本德中國有限公司)和自動化DNA提取設備抽提基因組DNA。利用眼科疾病基因檢測包混合引物(含381個基因,北京邁基諾基因科技有限責任公司合成),通過本科室測序平臺Illumina NextSeq 500進行基因測序。人類基因組參考序列版本選擇GRCh37(hg19)。
對二代測序突變結果進行Sanger測序法驗證。以患兒及其父母提取的基因組DNA為模板,使用Taq DNA聚合酶(立陶宛Fermentas公司)和引物(GeneTool軟件設計)對目標序列進行擴增。正向引物5′-TCCGCCCTGCCTGCTTCCTTCG-3′,反向引物5′-CAGGGCTGGCCGGGGAAGTAGG-3′,擴增片段長度為330堿基對(bp)。擴增條件為:95 ℃預變性3 min;95 ℃變性30 s,60 ℃退火30 s,72 ℃延伸45 s,重復循環35次;72 ℃終延伸10 min,25 ℃室溫保存。將聚合酶鏈反應(PCR)擴增的目的DNA片段純化后,用dGTPBigDye? Terminator(熒光標記終止物試劑盒,變性1 min;96 ℃10 s,50 ℃ 5 s,60 ℃ 4 min;重復循環25次;4 ℃保溫。采用乙醇/醋酸鈉純化,按BigDyeV3.1操作手冊進行。將純化產物變性后上測序儀(美國ABI公司)進行序列分析。
利用人類基因變異數據庫(HGMD)等工具查詢目標突變位點是否已被收錄。通過PubMed數據庫檢索是否有相關論文報道,被學術論文報道過的相關致病變異可確定為致病性突變。查詢ClinVar數據庫(https://www.ncbi.nlm.nih.gov/clinvar/)對本研究突變的收錄情況,確定人類基因變異和表型的關聯情況和證據。檢索基因組整合數據庫(gnomAD)以明確該突變在總人群中的出現頻率。Mutationt@sting蛋白預測軟件(http://mutationtaster.org/)預測插入突變對蛋白質功能的影響,以判斷其致病性。依據《遺傳變異分類標準與指南》[6]和2015年版的《ACMG Standard and Guidelines》[7]對本研究檢出突變的致病性進行判斷。
2 結果
Illumina測序平臺DNA測序發現,患兒PAX2基因存在c.70dupG(p.V26Gfs*28)雜合突變,導致PAX2基因的編碼蛋白轉錄因子配對盒2的第26位氨基酸由纈氨酸突變為甘氨酸并移碼28個密碼子后終止翻譯。正常表型的父母均不攜帶此變異。
Sanger測序法驗證發現,患兒及其父母的PAX2基因突變情況與二代測序一致(圖2)。患兒疾病表型推斷為PAX2基因c.70dupG(p.V26Gfs*28)雜合突變所致,系常染色體顯性遺傳。
圖2
PAX2基因c.70dupG(p.V26Gfs*28)突變位點Sanger測序驗證結果。2A.患兒;2B.患兒父親;2C. 患兒母親。患兒PAX2基因存在c.70dupG(p.V26Gfs*28)雜合突變,導致PAX2基因的編碼蛋白轉錄因子配對盒2的第26位氨基酸由纈氨酸突變為甘氨酸并移碼28個密碼子后終止翻譯(紅箭)
HGMD數據庫專業版和PubMed檢索發現PAX2基因c.70dupG(p.V26Gfs*28)突變致病性已有報道。人類基因變異和表型間關系的數據庫ClinVar亦有本研究中突變的致病性記錄,與局灶節段性腎小球硬化癥7型[OMIM:616002]、RCS[OMIM:120330]發病相關。利用Mutationt@sting蛋白質預測軟件對此位點進行預測分析,結果顯示為有害突變(disease causing),主要表現為移碼導致蛋白質翻譯提前終止而產生了截斷的無功能產物蛋白。根據《遺傳變異分類標準與指南》和2015年版《ACMG Standard and Guidelines》判斷該移碼突變為致病性變異,致病性證據非常強(等級PVS1)。gnomAD顯示該突變的dbSNP ID為rs768607170,僅在猶太人、非洲人和非芬蘭裔歐洲人中各出現過1例,人群總頻率約為十萬分之一;尚未在中國人或東亞人群中發現過。
2018年3月,患兒根據遺傳分析結果和建議在本院行針對性腎臟彩色超聲和腎功能檢查。腎臟彩色超聲檢查發現,雙腎大小形態正常;左腎實質可見大小約5.0 mm×4.8 mm囊性回聲,邊界清楚,后方回聲增強,左腎集合系統輕度分離,腎盂前后徑約8.0 mm;左腎小囊腫,雙腎彌漫性回聲改變。其余正常。腎功能檢查結果顯示,α1微球蛋白指標升高,其他各項指標在正常范圍內。提示患兒存在輕度腎臟異常或RCS綜合征腎臟病變的早期表現,需要定期復查及觀察。
3 討論
本例患兒眼部主要表現為右眼先天性白內障以及左眼視盤凹陷、發育不全、眼底“牽牛花綜合征”、視力持續下降等;腎部異常包括雙腎彌漫性回聲改變、左腎集合系統輕度分離、左腎小囊腫等特征性改變。患者這些臨床表型符合RCS的典型特征。研究者認為,RCS一般屬于PAX2基因突變引起的單基因遺傳病。根據孟德爾遺傳定律,本家系屬于常染色體顯性遺傳,針對常見的眼病遺傳病致病基因進行靶向捕獲和二代測序來篩查突變。測序分析發現患兒PAX2基因發生c.70dupG(p.V26Gfs*28)雜合突變導致RCS。
PAX2基因位于10q24.31,由13個外顯子組成,該基因的編碼蛋白轉錄因子配對盒2是轉錄因子基因家族的一員,它們的中心性特征為保守的DNA結合配對盒結構域。研究發現,PAX2基因突變可導致視覺神經組織缺損和腎臟發育不全,OMIM數據庫收錄PAX2基因為RCS[OMIM:120330]的致病基因。本研究檢出的PAX2基因c.70dupG(p.V26Gfs*28)雜合突變,在編碼鏈第70個核苷酸(第2外顯子)后插入了一個鳥苷酸(G)導致其產生了截斷的無功能蛋白產物。該患兒檢出的突變位于連續的7個鳥苷酸(G)這個突變熱點區域。Sanyanusin等[8]報道的PAX2基因c.76dupG(p.Val26Glyfs*28)突變和Thomas等[9]報道的PAX2基因c.69_70insG(G24fs*28)移碼突變與本例患兒PAX2基因c.70dupG(p.V26Gfs*28)的突變為同一突變。由于該突變所在位置連續的7個鳥苷酸(G)的特殊性,在檢索以往的研究文獻時需要注意該突變的書寫問題。
RCS有臨床表現異質性。相關眼部異常還可能包括角膜直徑小、視網膜缺損、鞏膜葡萄腫、視神經囊腫、小眼畸形和色素性黃斑發育不良等[1]。回顧PAX2基因突變和RCS的研究發現,本研究檢出突變的上下游10 bp區域內有多個已知致病性突變[10-13],推斷它可能為突變活躍區域。這些致病性突變在物理位置上相緊鄰,在蛋白結構中可能具有相近的作用。有一些PAX2基因突變導致的RCS患者伴隨CAKUT但沒有明顯的眼部異常,或不伴隨CAKUT但有眼部異常[14-15]。這種異質性不僅表現在不同家系的患者,甚至表現在相同基因突變的家系內部。Rieger[15]報道過一個RCS家系父子女3人表現出了很大的臨床表型差異。該家系中,父親表現為雙側視盤異常,最后因慢性腎炎死亡;兒子表現為黃斑和視網膜異常,但有正常的腎功能;女兒表現為正常眼,最后卻因進行性腎衰竭而死亡。本例患兒3月齡時因右眼先天性白內障而發現RCS眼部異常。經遺傳學分析,發現具有致病性的PAX2基因c.70dupG(p.V26Gfs*28)突變。根據遺傳學診斷結果,患兒經針對性的腎臟檢查發現雙腎彌漫性回聲改變、左腎集合系統輕度分離、左腎小囊腫等異常。遺傳學分析指導我們在該患兒腎臟出現臨床癥狀前發現了輕度異常或早期病變,這對于疾病的早期干預和治療具有重要意義。患兒右眼先天性白內障表型以往研究報道未見描述,這可能是PAX2基因突變導致的RCS綜合征臨床異質性的體現。
本研究從遺傳學角度分析了一例RCS患兒的致病原因,結合PAX2基因c.70dupG(p.V26Gfs*28)雜合突變的致病性和相關臨床檢查結果確認了一例RCS病例,該發現拓展了PAX2基因突變常染色體顯性遺傳RCS的表型譜,為理解該病的臨床表現和分子機制提供了有用信息。本研究PAX2基因致病突變的檢出和致病性分析表明,臨床異質性較大的遺傳性疾病的分子診斷對那些尚無明顯臨床表現的病變的早期發現及早期干預和治療具有重要意義。