引用本文: 尹小芳, 葉祖科, 湯秀容, 梁影影, 黎彥豪, 羅書科, 盧彥. Sorsby眼底營養不良一家系. 中華眼底病雜志, 2018, 34(6): 546-551. doi: 10.3760/cma.j.issn.1005-1015.2018.06.005 復制
Sorsby眼底營養不良(SFD)是一種常染色體顯性遺傳性疾病,發病率較低[1-2]。其通常于40~60歲發病,以黃斑區脈絡膜新生血管(CNV)及周邊視網膜脈絡膜萎縮而導致視力嚴重下降為主要特征[1]。目前國內尚未見SFD的詳細報道。我們在臨床中發現了一SFD家系,并對其進行了TIMP3基因外顯子測序驗證。現將結果報道如下。
1 對象和方法
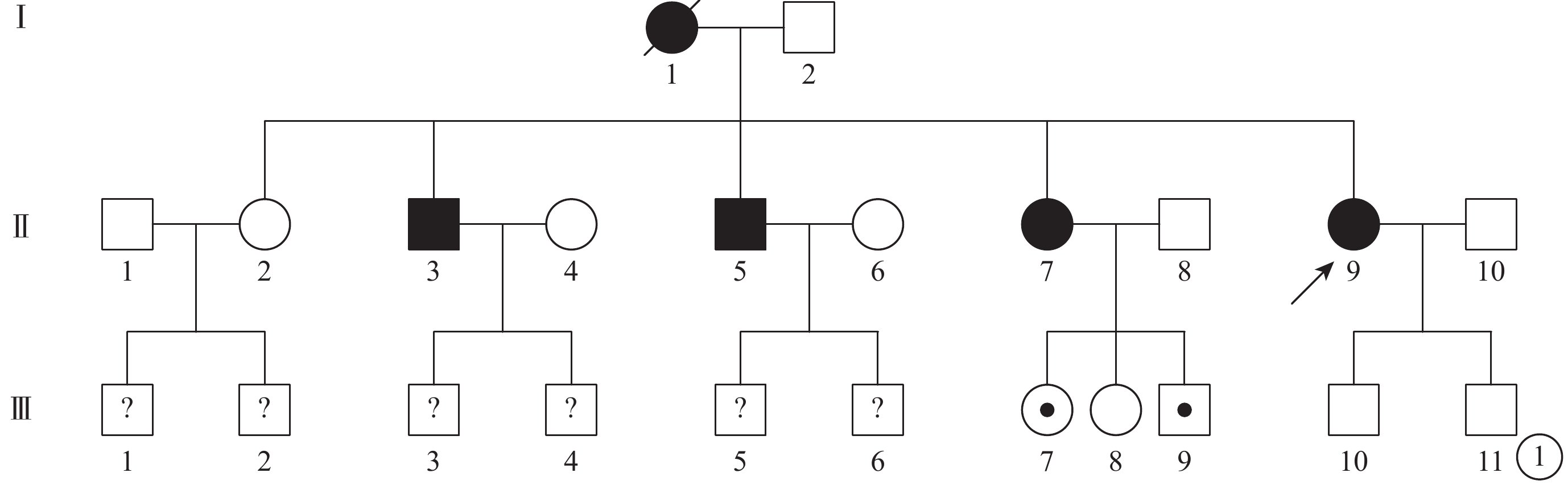
本研究獲得佛山市第二人民醫院醫學倫理委員會審核、批準,所有受試者均簽署知情同意書。2016年3~7月在我院進行SFD家系調查的一家系中4例患者及6名正常家系成員納入本研究。4例患者中,男女性各2例;6名成員中,男性4名,女性2名。家系成員年齡分別為44(Ⅱ9,先證者)、58(Ⅱ1)、56(Ⅱ3)、54(Ⅱ5)、46(Ⅱ7)、25(Ⅲ7)、23(Ⅲ8)、22(Ⅲ9)、21(Ⅲ10)、19(Ⅲ11)歲。采集完整的家系成員信息,繪制家系圖譜(圖1)。
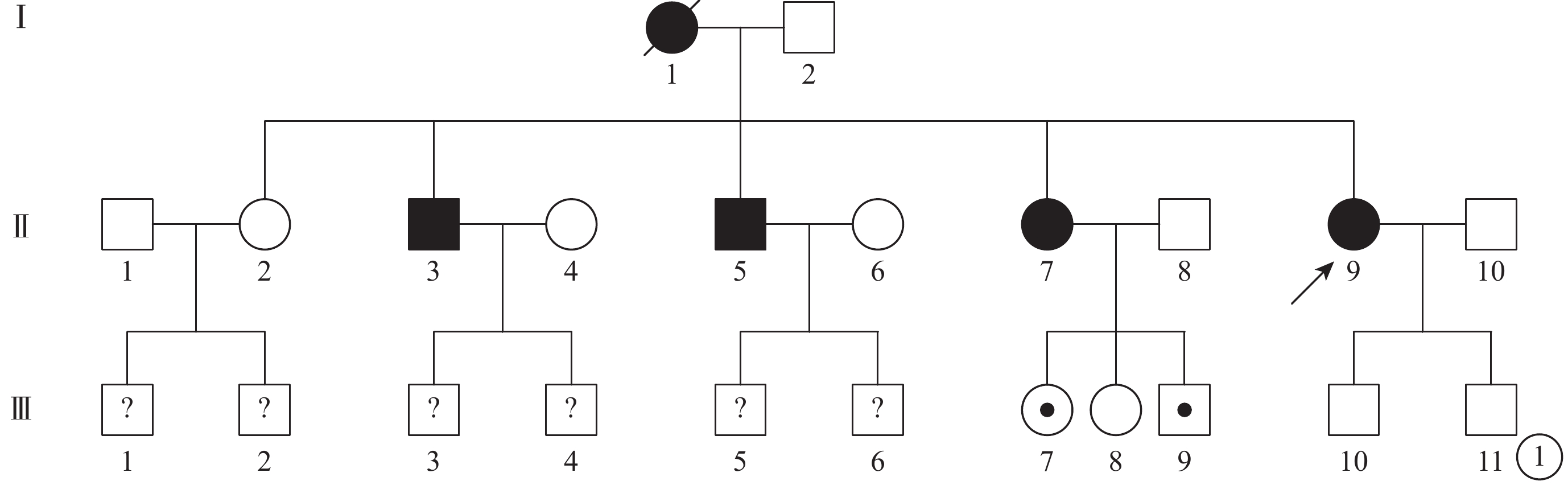 圖1
患者家系圖。■:男性患者;●女性患者;□:正常男性;○:正常女性;↑:先證者;
圖1
患者家系圖。■:男性患者;●女性患者;□:正常男性;○:正常女性;↑:先證者;
所有受試者均行最佳矯正視力(BCVA)、非接觸眼壓計、裂隙燈顯微鏡、間接檢眼鏡、眼底彩色照相及頻域光相干斷層掃描(SD-OCT)檢查。
采集所有受試者外周靜脈血2 ml,全血基因組DNA提取試劑盒(德國Qiagen公司)提取DNA并編號。參考基因組版本GRCh37/hg19,運用二代測序(NGS)方法對先證者進行眼科全套疾病相關外顯子捕獲檢測,并運用Sanger測序法針對發現的致病位點對其他9名受試者進行分離驗證(上海瑞立生物科技有限公司嘉興雅康博醫學檢驗所)。
2 結果
4例患者均在40~47歲出現明顯視力下降。其中,2例男性患者為雙眼發病,2例女性為單眼出現癥狀。據先證者口述,其母親(Ⅰ1)于40歲后先后出現雙眼視力明顯下降,曾在醫院診斷為“雙眼眼底病”,其余診療信息不詳。因家系中Ⅲ1、Ⅲ2、Ⅲ3、Ⅲ4、Ⅲ5、Ⅲ6均在外地,無法行眼部及血液學基因檢測。
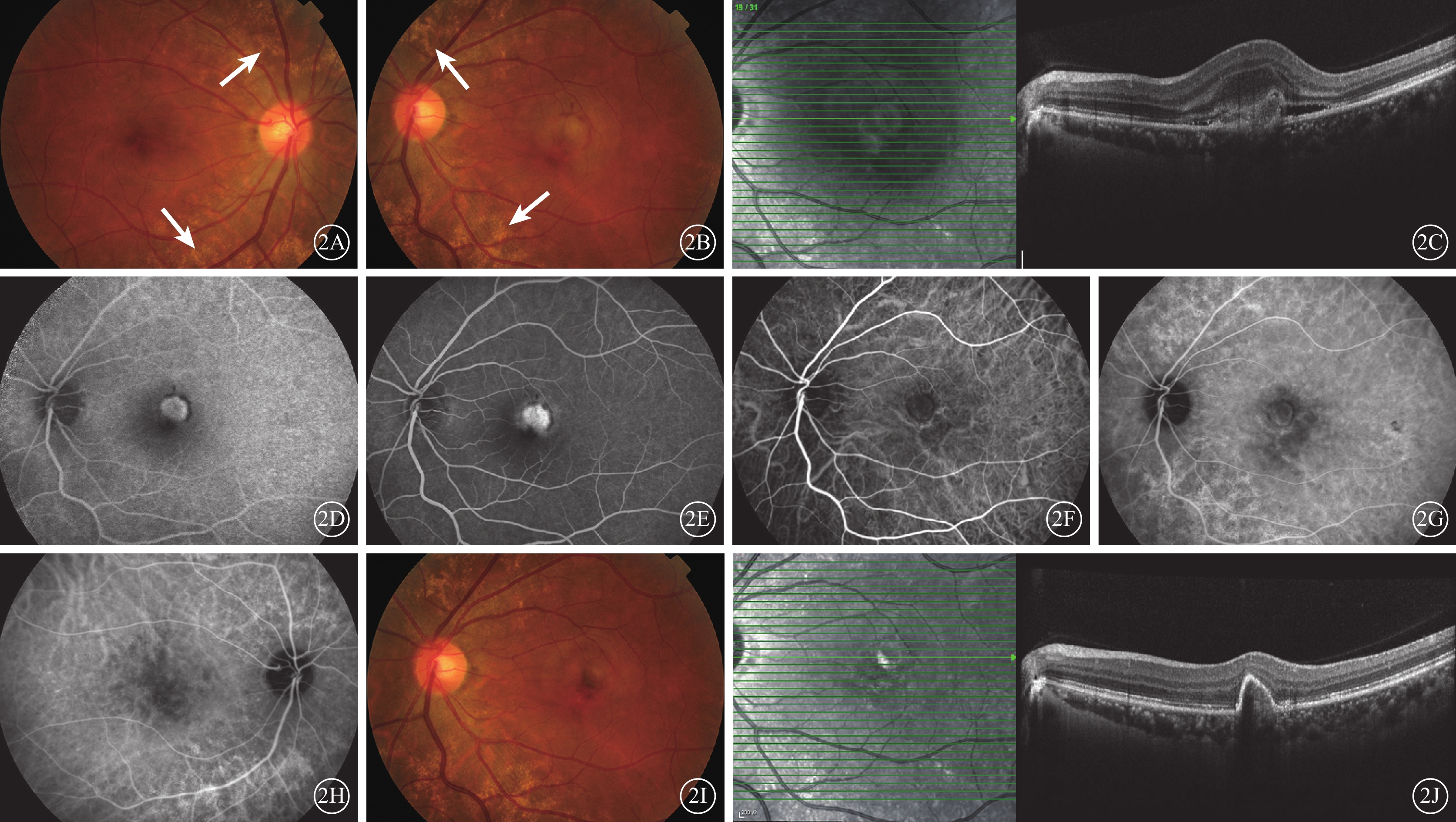
先證者(Ⅱ9),女,44歲。因左眼漸進性視力下降2個月就診。既往無眼部及全身病史。右眼、左眼BCVA分別為1.0、0.1;眼壓分別為10.9、12.1 mmHg(1 mmHg=0.133 kPa)。雙眼眼前節正常。雙眼視盤邊界清楚,顏色淡紅,上方、下方血管旁大量顆粒狀黃白色類玻璃膜疣沉積(圖2A,2B)。左眼黃斑中心及顳上方視網膜下黃白色隆起,伴出血水腫(圖2B)。SD-OCT檢查,左眼黃斑中心及顳上方視網膜色素上皮(RPE)結構破壞,可見不規則稍強反射信號隆起,邊界欠清,視網膜水腫增厚(圖2C);右眼未見明顯異常。熒光素眼底血管造影(FFA)檢查,左眼早期黃斑區可見脈絡膜新生血管(CNV),呈斑片狀強熒光、邊界清晰,隨時間延長,逐漸出現熒光素滲漏,其邊緣環繞出血遮蔽熒光(圖2D,2E);右眼視盤、黃斑及視網膜血管未見明顯異常熒光。吲哚青綠血管造影(ICGA)檢查,左眼早期黃斑區可見異常血管膜,邊界清晰,隨時間延長出現熒光素滲漏(圖2F,2G);雙眼黃斑區見脈絡膜毛細血管低灌注性弱熒光,視盤上方、下方大量斑塊狀稍強熒光、邊界清晰(圖2H)。全身檢查以及血尿常規、肝腎功能、紅細胞沉降率、C反應蛋白等實驗室輔助檢查未見異常。住院予以左眼玻璃體腔注射雷珠單抗(0.5 mg)治療。治療后1個月,左眼BCVA提高至0.6;SD-OCT檢查見黃斑水腫、CNV病灶較前明顯減退。給予左眼再次玻璃體腔注射雷珠單抗治療。治療后2個月,左眼黃斑區色素紊亂、小團灰白色隆起(圖2I)。SD-OCT檢查可見左眼黃斑水腫消退,RPE結構連續,CNV病灶縮小、穩定(圖2J)。左眼裸眼視力0.6(?1.00 DS/?0.50 DC×90°=1.0)。隨訪至今未見復發。
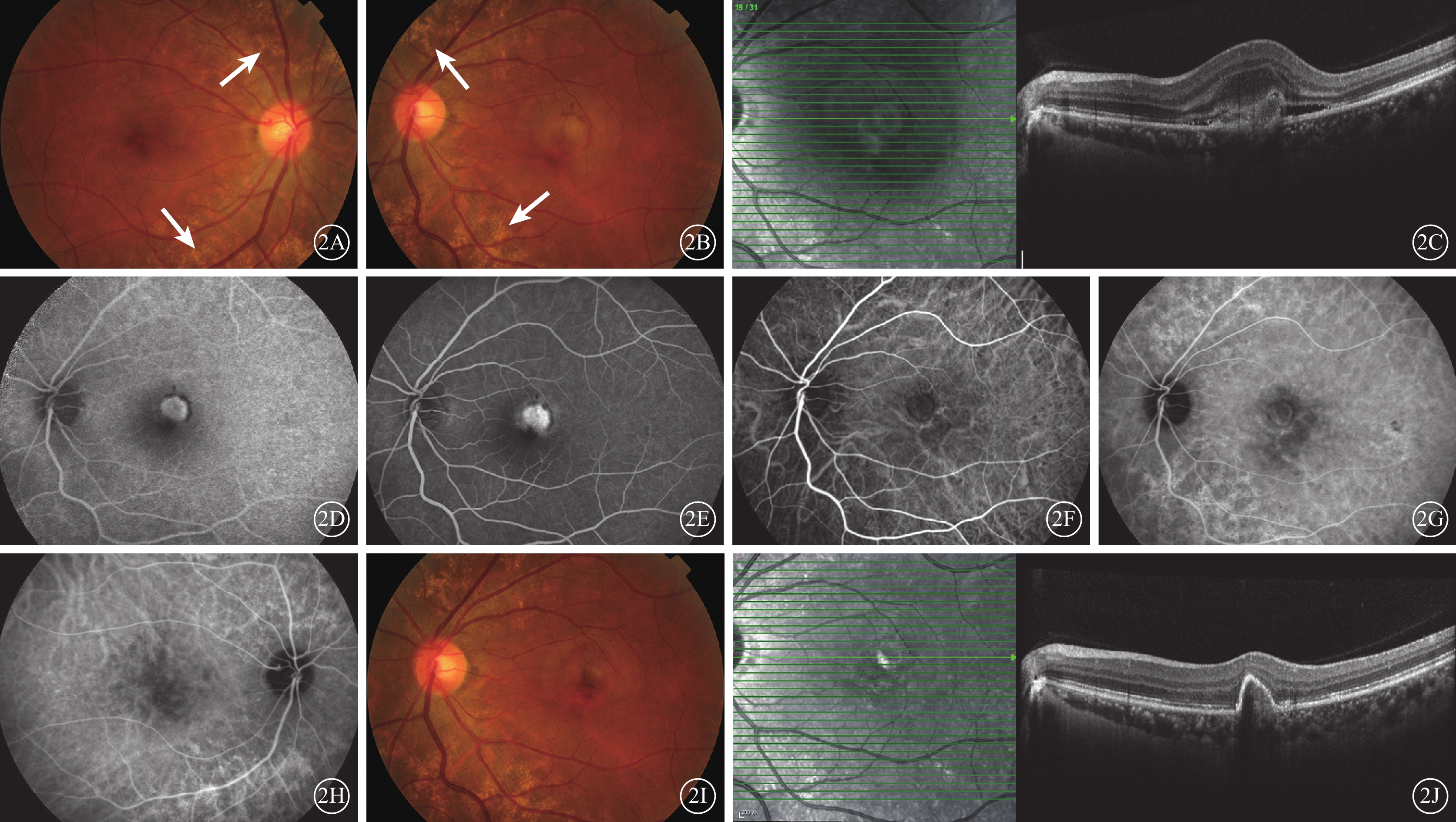 圖2
先證者(Ⅱ9)治療前后眼底檢查像。2A、2B.右眼、左眼彩色眼底像,雙眼可見視盤上方、下方血管旁大量顆粒狀黃白色類玻璃膜疣沉積(白箭),左眼黃斑中心及顳上方視網膜下黃白色隆起,視網膜出血、水腫;2C.左眼SD-OCT像,黃斑視網膜下RPE結構破壞,見不規則稍強反射信號隆起,邊界欠清,視網膜水腫增厚;2D.左眼3 min 49 s FFA像,黃斑區一斑片狀、邊界清晰的強熒光,周圍環繞出血遮蔽熒光;2E.左眼14 min 40 s FFA像,病灶熒光增強伴熒光素滲漏;2F.左眼49 s ICGA像,黃斑區邊界清晰的異常血管膜;2G.左眼8 min 27 s ICGA像,黃斑區病灶熒光素滲漏,病灶周圍脈絡膜血管呈低灌注性弱熒光,視盤上方及下方大量斑塊狀稍強熒光、邊界清晰;2H.右眼8 min 10 s ICGA像,黃斑區脈絡膜呈低灌注性弱熒光,視盤上方及下方大量邊界清晰的斑塊狀稍強熒光;2I.左眼治療后2個月彩色眼底像,黃斑區色素紊亂、小團灰白色隆起;2J.左眼治療后2個月SD-OCT像,CNV病灶穩定,RPE局部隆起、結構連續,視網膜未見水腫增厚
圖2
先證者(Ⅱ9)治療前后眼底檢查像。2A、2B.右眼、左眼彩色眼底像,雙眼可見視盤上方、下方血管旁大量顆粒狀黃白色類玻璃膜疣沉積(白箭),左眼黃斑中心及顳上方視網膜下黃白色隆起,視網膜出血、水腫;2C.左眼SD-OCT像,黃斑視網膜下RPE結構破壞,見不規則稍強反射信號隆起,邊界欠清,視網膜水腫增厚;2D.左眼3 min 49 s FFA像,黃斑區一斑片狀、邊界清晰的強熒光,周圍環繞出血遮蔽熒光;2E.左眼14 min 40 s FFA像,病灶熒光增強伴熒光素滲漏;2F.左眼49 s ICGA像,黃斑區邊界清晰的異常血管膜;2G.左眼8 min 27 s ICGA像,黃斑區病灶熒光素滲漏,病灶周圍脈絡膜血管呈低灌注性弱熒光,視盤上方及下方大量斑塊狀稍強熒光、邊界清晰;2H.右眼8 min 10 s ICGA像,黃斑區脈絡膜呈低灌注性弱熒光,視盤上方及下方大量邊界清晰的斑塊狀稍強熒光;2I.左眼治療后2個月彩色眼底像,黃斑區色素紊亂、小團灰白色隆起;2J.左眼治療后2個月SD-OCT像,CNV病灶穩定,RPE局部隆起、結構連續,視網膜未見水腫增厚
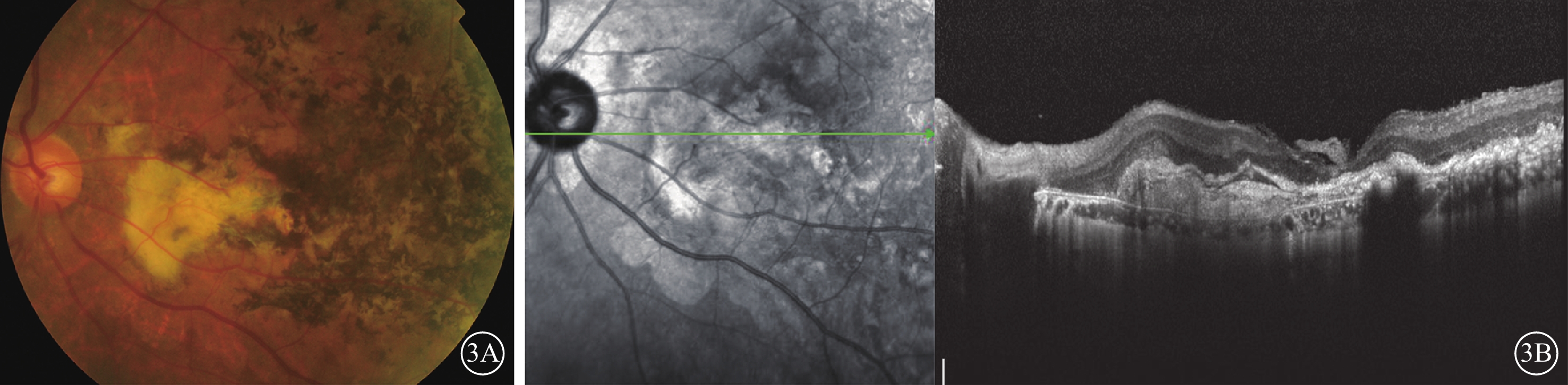
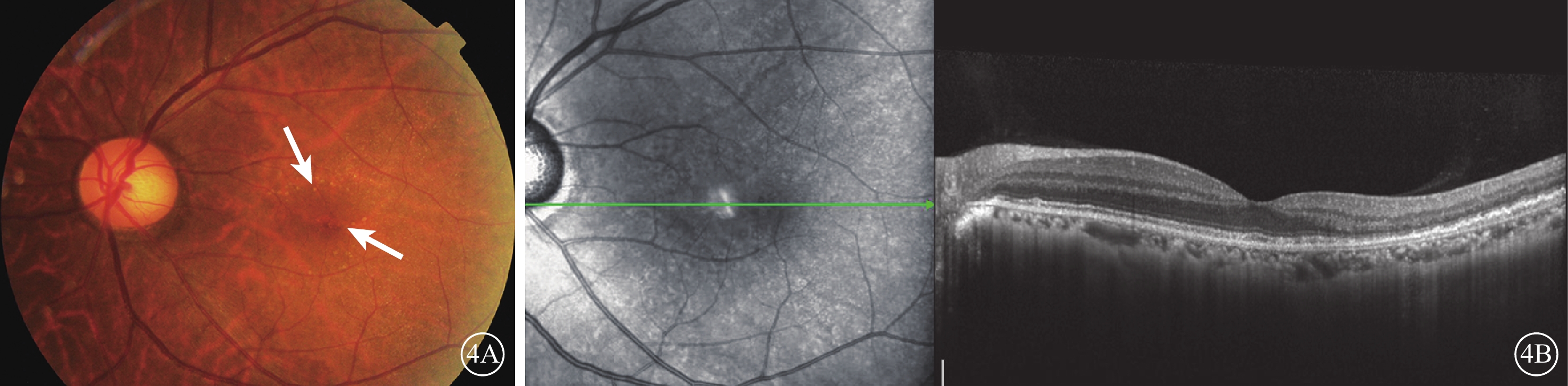
Ⅱ3患者10年前(46歲時)出現雙眼視力下降,9年前曾在我院以“雙眼CNV”先后兩次行雙眼光動力治療(PDT)。治療后雙眼視力無明顯提高。現隨先證者復診。右眼、左眼BCVA分別為0.05、0.1。雙眼眼前節未見明顯異常。雙眼視盤邊界清楚、顏色淡紅,黃斑區不規則斑塊狀黃白色瘢痕、萎縮灶,伴大量色素增生、沉著(圖3A)。SD-OCT檢查可見雙眼黃斑區RPE結構破壞,廣泛視網膜下團塊狀強反射信號隆起,視網膜局部可見增厚、層間弱反射腔隙(圖3B)。Ⅱ5患者7年前(47歲時)出現雙眼視力下降,呈進行性加重,既往診治經過不詳。雙眼BCVA 0.06。眼底表現及SD-OCT檢查與Ⅱ3高度相似。Ⅱ7患者6年前(40歲時)出現右眼視力下降,發病半年后在我院以“右眼CNV”行一次右眼PDT治療。右眼、左眼BCVA分別為0.03、0.5。右眼眼底表現及SD-OCT檢查類似Ⅱ3患者。左眼黃斑區周圍可見較多大小不等黃白色類玻璃膜疣樣沉著(圖4A)。SD-OCT檢查可見左眼黃斑顳側視網膜外核層萎縮變薄,RPE層不規則微隆起(圖4B)。
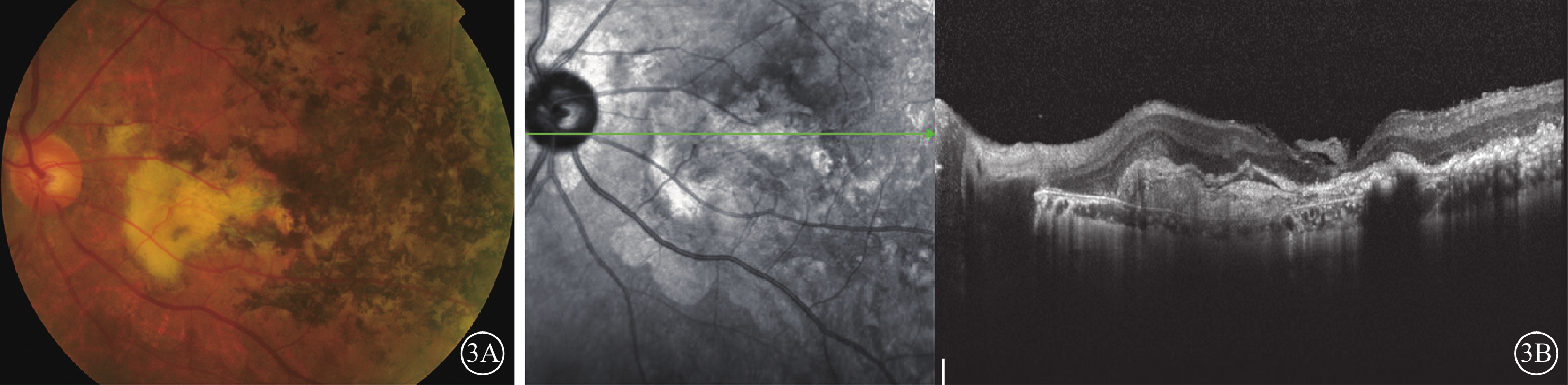 圖3
Ⅱ3患者左眼眼底檢查像。3A.彩色眼底像,黃斑區不規則斑塊狀黃白色瘢痕,伴大量色素增生、沉著;3B.SD-OCT像,黃斑區RPE結構破壞,視網膜下大量團塊狀強反射信號隆起,視網膜局部可見增厚、弱反射腔隙
圖3
Ⅱ3患者左眼眼底檢查像。3A.彩色眼底像,黃斑區不規則斑塊狀黃白色瘢痕,伴大量色素增生、沉著;3B.SD-OCT像,黃斑區RPE結構破壞,視網膜下大量團塊狀強反射信號隆起,視網膜局部可見增厚、弱反射腔隙
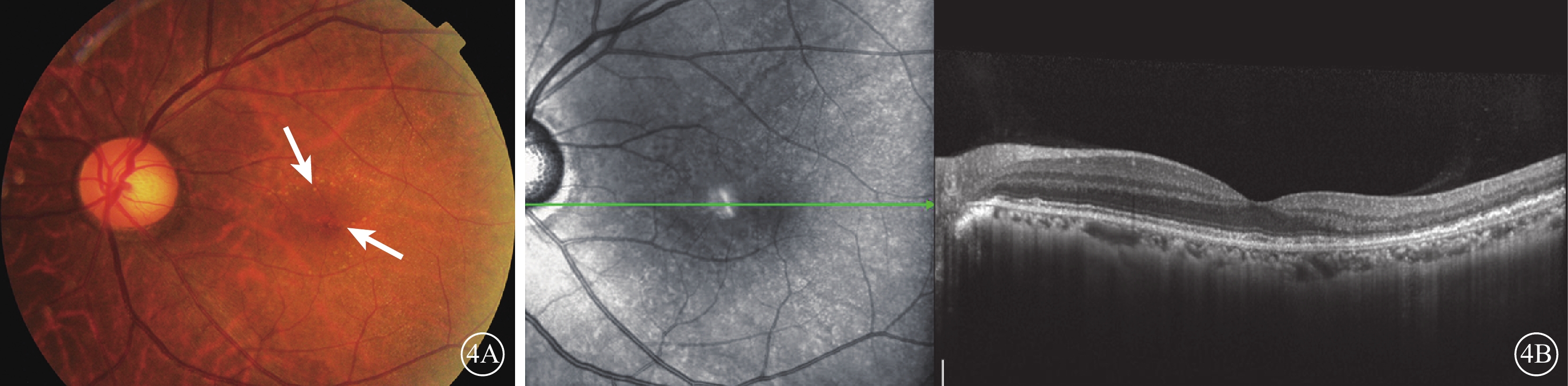 圖4
Ⅱ7患者左眼眼底檢查像。4A.彩色眼底像,黃斑周圍散在大小不等的黃白色顆粒狀類玻璃膜疣(白箭);4B.SD-OCT像,黃斑顳側視網膜外核層萎縮變薄,RPE層不規則微隆起
圖4
Ⅱ7患者左眼眼底檢查像。4A.彩色眼底像,黃斑周圍散在大小不等的黃白色顆粒狀類玻璃膜疣(白箭);4B.SD-OCT像,黃斑顳側視網膜外核層萎縮變薄,RPE層不規則微隆起
Ⅱ2、Ⅲ7、Ⅲ8、Ⅲ9、Ⅲ10、Ⅲ11受試者雙眼BCVA 1.0。除Ⅱ2受試者晶狀體輕度皮質性混濁外,其余受試者雙眼眼前節均未見明顯異常。6名受試者眼底彩色照相及SD-OCT檢查未見明顯異常改變。
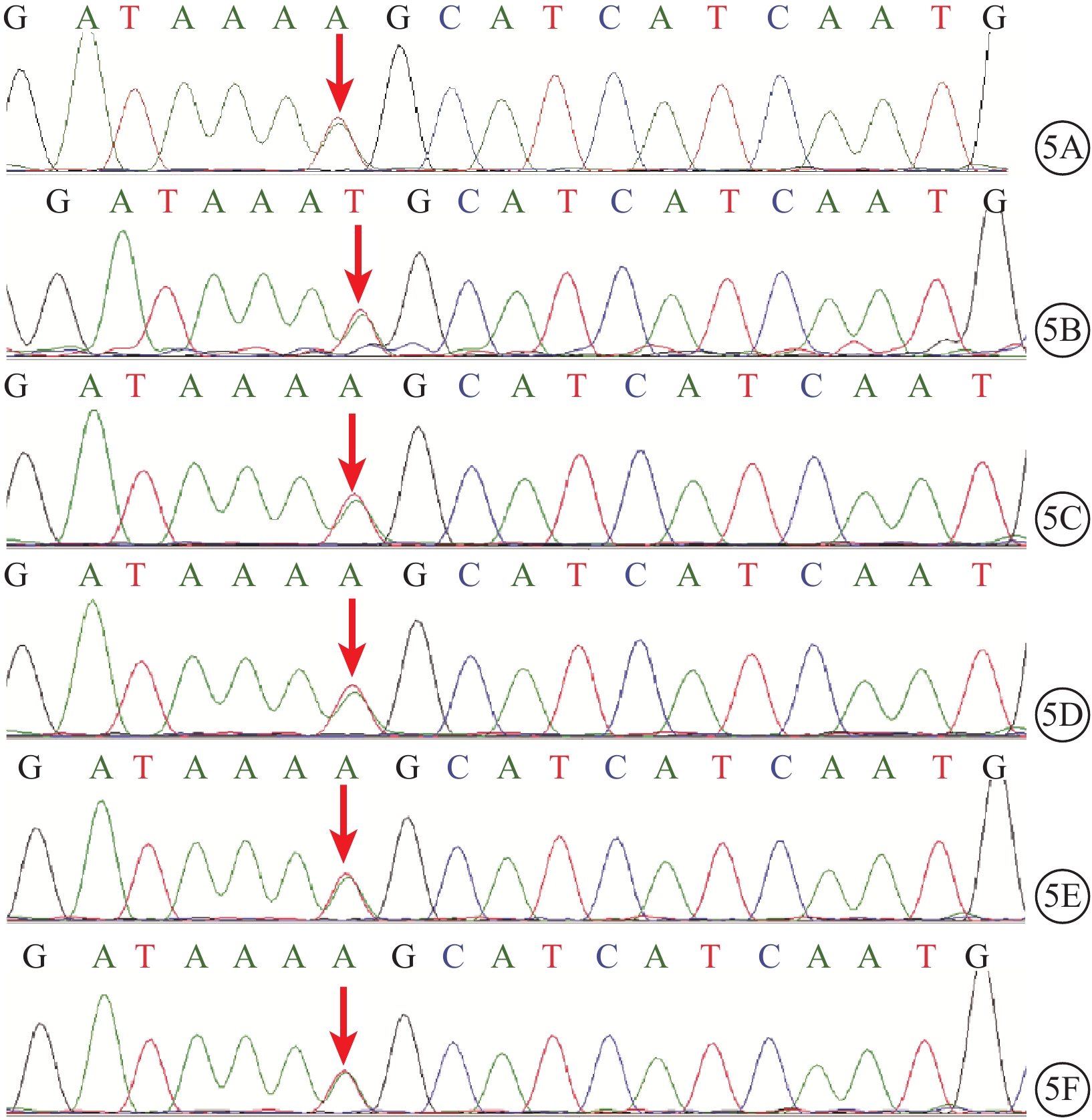
基因檢測結果顯示,先證者(Ⅱ9)的TIMP3基因第5號外顯子上存在c.610A>T堿基雜合突變,導致該基因編碼的第204位密碼子由絲氨酸變為半胱氨酸(TIMP3:NM_000362:Exon5:c.A610T/p.S204C)(圖5)。變異性質為Pathogenic/致病,遺傳方式為常染色體顯性遺傳。Sanger測序法驗證結果顯示,其他3例患者(Ⅱ3、Ⅱ5、Ⅱ7)及Ⅱ7患者的下一代Ⅲ7、Ⅲ9受試者在該位點均檢測到與先證者相同的變異。
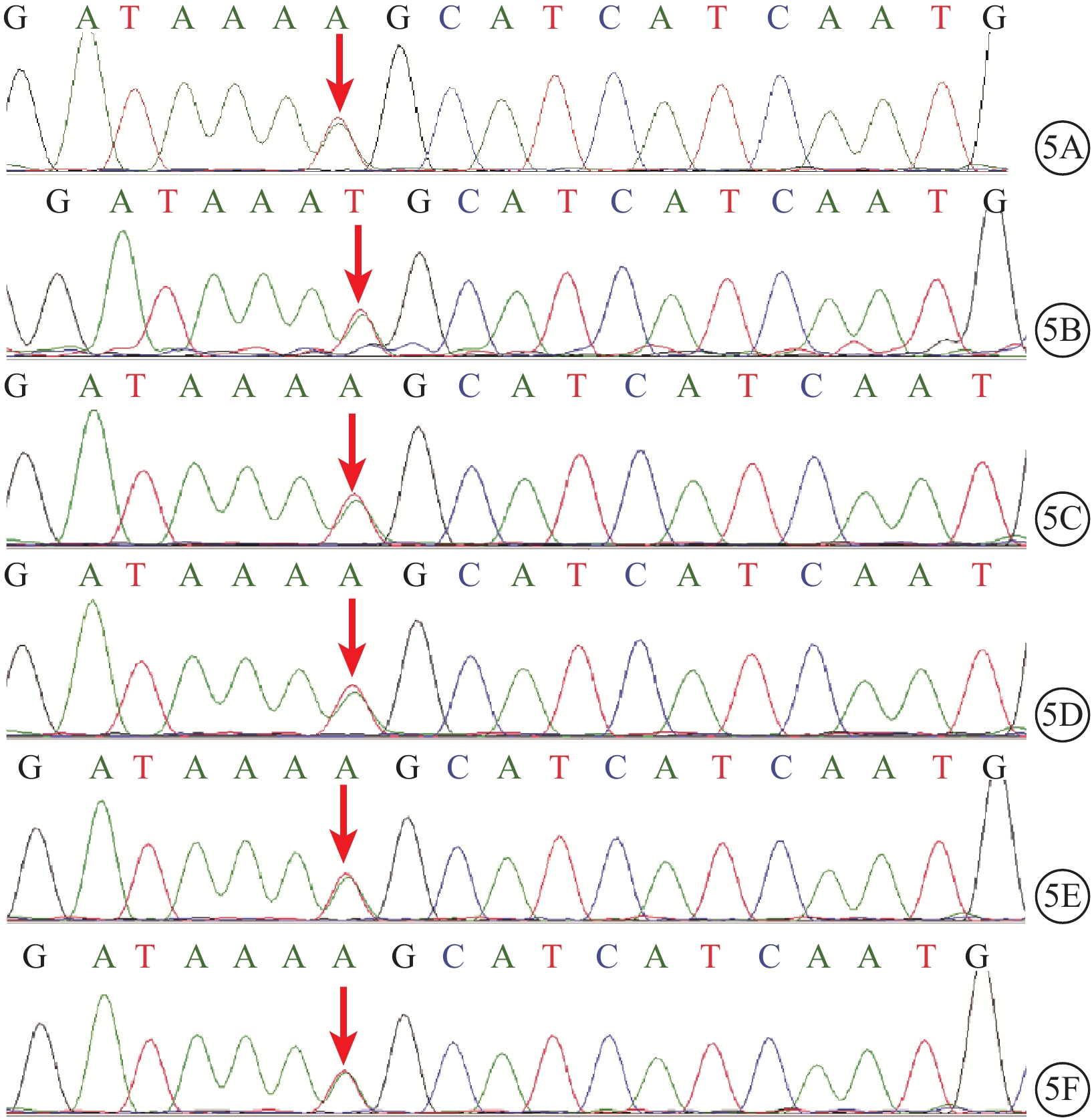 圖5
基因二代測序圖。5A~5F.先證者、Ⅱ3、Ⅱ5、Ⅱ7、Ⅲ7及Ⅲ9。TIMP3基因Chr22:33255338,第5號外顯子上存在c.610A>T堿基雜合突變(紅箭),導致該基因編碼的第204位密碼子由絲氨酸變為半胱氨酸
圖5
基因二代測序圖。5A~5F.先證者、Ⅱ3、Ⅱ5、Ⅱ7、Ⅲ7及Ⅲ9。TIMP3基因Chr22:33255338,第5號外顯子上存在c.610A>T堿基雜合突變(紅箭),導致該基因編碼的第204位密碼子由絲氨酸變為半胱氨酸
3 討論
SFD可雙眼同時或先后發病,以中心視力明顯下降為主要特征,可伴視物變形、夜盲癥、閃光感[3]。病程早期眼底常可見類玻璃膜疣樣黃白色顆粒狀沉積,其形態與玻璃膜疣類似,位于RPE下[4]。隨病程進展,SFD主要表現為以下兩種形式[5]。(1)黃白色類玻璃膜疣沉積物逐漸融合,出現黃斑CNV伴出血、水腫、滲出,引起視力急劇下降;晚期黃斑萎縮、瘢痕形成。(2)黃斑區脈絡膜毛細血管血供不足導致的進行性RPE萎縮,類似于地圖狀萎縮,引起中心視力進行性下降。病變也可向周邊發展,出現周邊內層脈絡膜和外層視網膜萎縮,導致周邊視野損害、夜盲癥。患者視力預后較差,通常介于數指至0.1之間[6]。由于SFD眼底表現與特發性CNV、老年性黃斑變性相似,臨床上容易誤診、漏診。明確的家族史是鑒別診斷中最重要的一點。本家系患者可見典型的黃斑區CNV、后極部斑點狀類玻璃膜疣沉積或局部外層視網膜萎縮等表現,且先證者ICGA檢查可見黃斑區脈絡膜低灌注性弱熒光,與文獻報道相符[5]。本家系患者有明確家族史,臨床表現典型且處于疾病不同階段,診斷明確。
SFD的發病機制尚未完全明確。現有研究表明,22號染色體上的TIMP3基因突變與SFD密切相關[7-10]。迄今為止已經發現18個不同的TIMP3基因突變與SFD有關,除1個位于內含子4與外顯子連接處外,其余17個突變位點均位于5號外顯子上(Cys24Arg、Ser38Cys、Tyr151Cys、Glu162Lys、Glu162STOP、Tyr174Cys、Tyr177Cys、Ser179Cys、His181Arg、Tyr182Cys、Gly189Cys、Gly190Cys、Tyr191Cys、Ser193Cys、Tyr195Cys、Trp198Cys和Ser204Cys)[7-21]。本家系4例患者經二代測序法檢測均發現TIMP3基因5號外顯子上c.610A>T,導致204密碼子編碼的氨基酸由絲氨酸變成半胱氨酸,即Ser204Cys突變型。TIMP3是基質金屬蛋白酶(MMP)抑制蛋白家族成員TIMP之一,是一種鋅依賴的蛋白水解酶。TIMP通過抑制MMP活性來調節細胞外基質的形成,從而在調節細胞生長、遷移、轉化、凋亡及血管化等方面起著重要作用。正常情況下,視網膜的TIMP3主要由RPE細胞分泌到Bruch膜中。TIMP3基因的突變可能導致了大量異常TIMP3在Bruch膜上的堆積,同時減弱了TIMP3對MMP的抑制作用,刺激血管的形成、產生CNV[22-23]。
引起SFD患者視力下降的最主要原因是黃斑CNV形成,因此目前關于SFD的治療多集中于此。傳統氬激光治療效果欠佳,容易引起CNV復發[24]。研究發現,SFD患者CNV進行PDT治療的次數要比滲出型老年性黃斑變性更多,SFD的CNV更難封閉、滲漏更難消退[25]。PDT的遠期效果不理想,對視力的提高、病灶范圍的縮小幫助不大[25]。此外,有研究顯示PDT聯合玻璃體腔注射曲安奈德短期內可獲得較好療效[26]。抗血管內皮生長因子(VEGF)藥物作為目前CNV的一線治療,在SFD患者中的應用已取得良好效果[16, 27]。抗VEGF藥物治療雖然不能阻止CNV復發,但可以減小其瘢痕化的范圍、改善預后[28]。也有研究認為短效的抗VEGF藥物與長效激素治療相結合可能是治療SFD患者CNV的一種有效選擇[25]。本家系中Ⅱ3和Ⅱ7患者經PDT治療后視力改善不明顯,黃斑區形成大面積瘢痕及色素增生。先證者(Ⅱ9)經過兩次雷珠單抗治療后矯正視力恢復正常,隨訪兩年未見明顯復發跡象。值得注意的是,該患者ICGA檢查可見黃斑區脈絡膜血管低灌注狀態,黃斑外層相對血供不足,抗VEGF藥物治療雖然能短期內抑制CNV生長、減少其滲漏,但遠期效果有待于進一步觀察。
本家系中Ⅱ7患者及先證者(Ⅱ9)雖然目前單眼出現癥狀、嚴重影響視力,但其對側眼均出現了黃白色類玻璃膜疣沉積及黃斑周圍視網膜外層萎縮(Ⅱ7患者)。這可以視為兩只眼處于發病的不同階段,即一只眼有典型的眼底改變,另一只眼處于病程早期,可能在之后的某個人生階段中發病。遺憾的是該病除了定期隨訪,當前并無明確預防措施。基因治療可能是SFD未來的一大方向,但這尚需要進一步研究。重組TIMP3基因可以從根本上治療SFD。對TIMP3基因突變陽性的基因攜帶者(如本家系Ⅲ7、Ⅲ9),在出現病癥前如能進行基因治療,有可能最大程度地保留患者的視力。
Sorsby眼底營養不良(SFD)是一種常染色體顯性遺傳性疾病,發病率較低[1-2]。其通常于40~60歲發病,以黃斑區脈絡膜新生血管(CNV)及周邊視網膜脈絡膜萎縮而導致視力嚴重下降為主要特征[1]。目前國內尚未見SFD的詳細報道。我們在臨床中發現了一SFD家系,并對其進行了TIMP3基因外顯子測序驗證。現將結果報道如下。
1 對象和方法
本研究獲得佛山市第二人民醫院醫學倫理委員會審核、批準,所有受試者均簽署知情同意書。2016年3~7月在我院進行SFD家系調查的一家系中4例患者及6名正常家系成員納入本研究。4例患者中,男女性各2例;6名成員中,男性4名,女性2名。家系成員年齡分別為44(Ⅱ9,先證者)、58(Ⅱ1)、56(Ⅱ3)、54(Ⅱ5)、46(Ⅱ7)、25(Ⅲ7)、23(Ⅲ8)、22(Ⅲ9)、21(Ⅲ10)、19(Ⅲ11)歲。采集完整的家系成員信息,繪制家系圖譜(圖1)。
圖1
患者家系圖。■:男性患者;●女性患者;□:正常男性;○:正常女性;↑:先證者;
所有受試者均行最佳矯正視力(BCVA)、非接觸眼壓計、裂隙燈顯微鏡、間接檢眼鏡、眼底彩色照相及頻域光相干斷層掃描(SD-OCT)檢查。
采集所有受試者外周靜脈血2 ml,全血基因組DNA提取試劑盒(德國Qiagen公司)提取DNA并編號。參考基因組版本GRCh37/hg19,運用二代測序(NGS)方法對先證者進行眼科全套疾病相關外顯子捕獲檢測,并運用Sanger測序法針對發現的致病位點對其他9名受試者進行分離驗證(上海瑞立生物科技有限公司嘉興雅康博醫學檢驗所)。
2 結果
4例患者均在40~47歲出現明顯視力下降。其中,2例男性患者為雙眼發病,2例女性為單眼出現癥狀。據先證者口述,其母親(Ⅰ1)于40歲后先后出現雙眼視力明顯下降,曾在醫院診斷為“雙眼眼底病”,其余診療信息不詳。因家系中Ⅲ1、Ⅲ2、Ⅲ3、Ⅲ4、Ⅲ5、Ⅲ6均在外地,無法行眼部及血液學基因檢測。
先證者(Ⅱ9),女,44歲。因左眼漸進性視力下降2個月就診。既往無眼部及全身病史。右眼、左眼BCVA分別為1.0、0.1;眼壓分別為10.9、12.1 mmHg(1 mmHg=0.133 kPa)。雙眼眼前節正常。雙眼視盤邊界清楚,顏色淡紅,上方、下方血管旁大量顆粒狀黃白色類玻璃膜疣沉積(圖2A,2B)。左眼黃斑中心及顳上方視網膜下黃白色隆起,伴出血水腫(圖2B)。SD-OCT檢查,左眼黃斑中心及顳上方視網膜色素上皮(RPE)結構破壞,可見不規則稍強反射信號隆起,邊界欠清,視網膜水腫增厚(圖2C);右眼未見明顯異常。熒光素眼底血管造影(FFA)檢查,左眼早期黃斑區可見脈絡膜新生血管(CNV),呈斑片狀強熒光、邊界清晰,隨時間延長,逐漸出現熒光素滲漏,其邊緣環繞出血遮蔽熒光(圖2D,2E);右眼視盤、黃斑及視網膜血管未見明顯異常熒光。吲哚青綠血管造影(ICGA)檢查,左眼早期黃斑區可見異常血管膜,邊界清晰,隨時間延長出現熒光素滲漏(圖2F,2G);雙眼黃斑區見脈絡膜毛細血管低灌注性弱熒光,視盤上方、下方大量斑塊狀稍強熒光、邊界清晰(圖2H)。全身檢查以及血尿常規、肝腎功能、紅細胞沉降率、C反應蛋白等實驗室輔助檢查未見異常。住院予以左眼玻璃體腔注射雷珠單抗(0.5 mg)治療。治療后1個月,左眼BCVA提高至0.6;SD-OCT檢查見黃斑水腫、CNV病灶較前明顯減退。給予左眼再次玻璃體腔注射雷珠單抗治療。治療后2個月,左眼黃斑區色素紊亂、小團灰白色隆起(圖2I)。SD-OCT檢查可見左眼黃斑水腫消退,RPE結構連續,CNV病灶縮小、穩定(圖2J)。左眼裸眼視力0.6(?1.00 DS/?0.50 DC×90°=1.0)。隨訪至今未見復發。
圖2
先證者(Ⅱ9)治療前后眼底檢查像。2A、2B.右眼、左眼彩色眼底像,雙眼可見視盤上方、下方血管旁大量顆粒狀黃白色類玻璃膜疣沉積(白箭),左眼黃斑中心及顳上方視網膜下黃白色隆起,視網膜出血、水腫;2C.左眼SD-OCT像,黃斑視網膜下RPE結構破壞,見不規則稍強反射信號隆起,邊界欠清,視網膜水腫增厚;2D.左眼3 min 49 s FFA像,黃斑區一斑片狀、邊界清晰的強熒光,周圍環繞出血遮蔽熒光;2E.左眼14 min 40 s FFA像,病灶熒光增強伴熒光素滲漏;2F.左眼49 s ICGA像,黃斑區邊界清晰的異常血管膜;2G.左眼8 min 27 s ICGA像,黃斑區病灶熒光素滲漏,病灶周圍脈絡膜血管呈低灌注性弱熒光,視盤上方及下方大量斑塊狀稍強熒光、邊界清晰;2H.右眼8 min 10 s ICGA像,黃斑區脈絡膜呈低灌注性弱熒光,視盤上方及下方大量邊界清晰的斑塊狀稍強熒光;2I.左眼治療后2個月彩色眼底像,黃斑區色素紊亂、小團灰白色隆起;2J.左眼治療后2個月SD-OCT像,CNV病灶穩定,RPE局部隆起、結構連續,視網膜未見水腫增厚
Ⅱ3患者10年前(46歲時)出現雙眼視力下降,9年前曾在我院以“雙眼CNV”先后兩次行雙眼光動力治療(PDT)。治療后雙眼視力無明顯提高。現隨先證者復診。右眼、左眼BCVA分別為0.05、0.1。雙眼眼前節未見明顯異常。雙眼視盤邊界清楚、顏色淡紅,黃斑區不規則斑塊狀黃白色瘢痕、萎縮灶,伴大量色素增生、沉著(圖3A)。SD-OCT檢查可見雙眼黃斑區RPE結構破壞,廣泛視網膜下團塊狀強反射信號隆起,視網膜局部可見增厚、層間弱反射腔隙(圖3B)。Ⅱ5患者7年前(47歲時)出現雙眼視力下降,呈進行性加重,既往診治經過不詳。雙眼BCVA 0.06。眼底表現及SD-OCT檢查與Ⅱ3高度相似。Ⅱ7患者6年前(40歲時)出現右眼視力下降,發病半年后在我院以“右眼CNV”行一次右眼PDT治療。右眼、左眼BCVA分別為0.03、0.5。右眼眼底表現及SD-OCT檢查類似Ⅱ3患者。左眼黃斑區周圍可見較多大小不等黃白色類玻璃膜疣樣沉著(圖4A)。SD-OCT檢查可見左眼黃斑顳側視網膜外核層萎縮變薄,RPE層不規則微隆起(圖4B)。
圖3
Ⅱ3患者左眼眼底檢查像。3A.彩色眼底像,黃斑區不規則斑塊狀黃白色瘢痕,伴大量色素增生、沉著;3B.SD-OCT像,黃斑區RPE結構破壞,視網膜下大量團塊狀強反射信號隆起,視網膜局部可見增厚、弱反射腔隙
圖4
Ⅱ7患者左眼眼底檢查像。4A.彩色眼底像,黃斑周圍散在大小不等的黃白色顆粒狀類玻璃膜疣(白箭);4B.SD-OCT像,黃斑顳側視網膜外核層萎縮變薄,RPE層不規則微隆起
Ⅱ2、Ⅲ7、Ⅲ8、Ⅲ9、Ⅲ10、Ⅲ11受試者雙眼BCVA 1.0。除Ⅱ2受試者晶狀體輕度皮質性混濁外,其余受試者雙眼眼前節均未見明顯異常。6名受試者眼底彩色照相及SD-OCT檢查未見明顯異常改變。
基因檢測結果顯示,先證者(Ⅱ9)的TIMP3基因第5號外顯子上存在c.610A>T堿基雜合突變,導致該基因編碼的第204位密碼子由絲氨酸變為半胱氨酸(TIMP3:NM_000362:Exon5:c.A610T/p.S204C)(圖5)。變異性質為Pathogenic/致病,遺傳方式為常染色體顯性遺傳。Sanger測序法驗證結果顯示,其他3例患者(Ⅱ3、Ⅱ5、Ⅱ7)及Ⅱ7患者的下一代Ⅲ7、Ⅲ9受試者在該位點均檢測到與先證者相同的變異。
圖5
基因二代測序圖。5A~5F.先證者、Ⅱ3、Ⅱ5、Ⅱ7、Ⅲ7及Ⅲ9。TIMP3基因Chr22:33255338,第5號外顯子上存在c.610A>T堿基雜合突變(紅箭),導致該基因編碼的第204位密碼子由絲氨酸變為半胱氨酸
3 討論
SFD可雙眼同時或先后發病,以中心視力明顯下降為主要特征,可伴視物變形、夜盲癥、閃光感[3]。病程早期眼底常可見類玻璃膜疣樣黃白色顆粒狀沉積,其形態與玻璃膜疣類似,位于RPE下[4]。隨病程進展,SFD主要表現為以下兩種形式[5]。(1)黃白色類玻璃膜疣沉積物逐漸融合,出現黃斑CNV伴出血、水腫、滲出,引起視力急劇下降;晚期黃斑萎縮、瘢痕形成。(2)黃斑區脈絡膜毛細血管血供不足導致的進行性RPE萎縮,類似于地圖狀萎縮,引起中心視力進行性下降。病變也可向周邊發展,出現周邊內層脈絡膜和外層視網膜萎縮,導致周邊視野損害、夜盲癥。患者視力預后較差,通常介于數指至0.1之間[6]。由于SFD眼底表現與特發性CNV、老年性黃斑變性相似,臨床上容易誤診、漏診。明確的家族史是鑒別診斷中最重要的一點。本家系患者可見典型的黃斑區CNV、后極部斑點狀類玻璃膜疣沉積或局部外層視網膜萎縮等表現,且先證者ICGA檢查可見黃斑區脈絡膜低灌注性弱熒光,與文獻報道相符[5]。本家系患者有明確家族史,臨床表現典型且處于疾病不同階段,診斷明確。
SFD的發病機制尚未完全明確。現有研究表明,22號染色體上的TIMP3基因突變與SFD密切相關[7-10]。迄今為止已經發現18個不同的TIMP3基因突變與SFD有關,除1個位于內含子4與外顯子連接處外,其余17個突變位點均位于5號外顯子上(Cys24Arg、Ser38Cys、Tyr151Cys、Glu162Lys、Glu162STOP、Tyr174Cys、Tyr177Cys、Ser179Cys、His181Arg、Tyr182Cys、Gly189Cys、Gly190Cys、Tyr191Cys、Ser193Cys、Tyr195Cys、Trp198Cys和Ser204Cys)[7-21]。本家系4例患者經二代測序法檢測均發現TIMP3基因5號外顯子上c.610A>T,導致204密碼子編碼的氨基酸由絲氨酸變成半胱氨酸,即Ser204Cys突變型。TIMP3是基質金屬蛋白酶(MMP)抑制蛋白家族成員TIMP之一,是一種鋅依賴的蛋白水解酶。TIMP通過抑制MMP活性來調節細胞外基質的形成,從而在調節細胞生長、遷移、轉化、凋亡及血管化等方面起著重要作用。正常情況下,視網膜的TIMP3主要由RPE細胞分泌到Bruch膜中。TIMP3基因的突變可能導致了大量異常TIMP3在Bruch膜上的堆積,同時減弱了TIMP3對MMP的抑制作用,刺激血管的形成、產生CNV[22-23]。
引起SFD患者視力下降的最主要原因是黃斑CNV形成,因此目前關于SFD的治療多集中于此。傳統氬激光治療效果欠佳,容易引起CNV復發[24]。研究發現,SFD患者CNV進行PDT治療的次數要比滲出型老年性黃斑變性更多,SFD的CNV更難封閉、滲漏更難消退[25]。PDT的遠期效果不理想,對視力的提高、病灶范圍的縮小幫助不大[25]。此外,有研究顯示PDT聯合玻璃體腔注射曲安奈德短期內可獲得較好療效[26]。抗血管內皮生長因子(VEGF)藥物作為目前CNV的一線治療,在SFD患者中的應用已取得良好效果[16, 27]。抗VEGF藥物治療雖然不能阻止CNV復發,但可以減小其瘢痕化的范圍、改善預后[28]。也有研究認為短效的抗VEGF藥物與長效激素治療相結合可能是治療SFD患者CNV的一種有效選擇[25]。本家系中Ⅱ3和Ⅱ7患者經PDT治療后視力改善不明顯,黃斑區形成大面積瘢痕及色素增生。先證者(Ⅱ9)經過兩次雷珠單抗治療后矯正視力恢復正常,隨訪兩年未見明顯復發跡象。值得注意的是,該患者ICGA檢查可見黃斑區脈絡膜血管低灌注狀態,黃斑外層相對血供不足,抗VEGF藥物治療雖然能短期內抑制CNV生長、減少其滲漏,但遠期效果有待于進一步觀察。
本家系中Ⅱ7患者及先證者(Ⅱ9)雖然目前單眼出現癥狀、嚴重影響視力,但其對側眼均出現了黃白色類玻璃膜疣沉積及黃斑周圍視網膜外層萎縮(Ⅱ7患者)。這可以視為兩只眼處于發病的不同階段,即一只眼有典型的眼底改變,另一只眼處于病程早期,可能在之后的某個人生階段中發病。遺憾的是該病除了定期隨訪,當前并無明確預防措施。基因治療可能是SFD未來的一大方向,但這尚需要進一步研究。重組TIMP3基因可以從根本上治療SFD。對TIMP3基因突變陽性的基因攜帶者(如本家系Ⅲ7、Ⅲ9),在出現病癥前如能進行基因治療,有可能最大程度地保留患者的視力。