引用本文: 王曉光, 劉海軍, 張少弛, 齊小龍, 潘波, 莊文娟, 盛迅倫. 視網膜色素變性和視錐-視桿細胞營養不良患者的基因型及臨床表型分析. 中華眼底病雜志, 2018, 34(6): 526-535. doi: 10.3760/cma.j.issn.1005-1015.2018.06.002 復制
視網膜色素變性(RP)和視錐-視桿細胞營養不良(CORD)是臨床上最常見的致盲性遺傳性視網膜疾病(IRD),在早期因缺乏特征性眼部表現時極易漏診和誤診[1-2]。由于兩者具有高度遺傳異質性和臨床異質性[3-5],眾多基因缺陷均可導致上述兩種疾病的發生,尚不能通過基因檢測結果直接進行準確診斷,限制了本病的基因診斷在臨床的廣泛應用。本研究應用目標序列捕獲結合二代測序技術對RP和CORD患者進行致病基因突變檢測,同時結合臨床表型分析,初步探討RP和CORD的診斷和鑒別診斷。現將結果報道如下。
1 對象和方法
2015年9月至2017年4月在寧夏眼科醫院檢查確診的RP患者37例及其三代以內家庭成員84名和CORD患者6例及其三代以內家庭成員11名納入研究。對照組為300名與受檢者無親緣關系的健康成年人。本研究經寧夏回族自治區人民醫院倫理委員會審核批準,遵循赫爾辛基宣言,所有受檢者和未成年患者監護人均簽署知情同意書。
參照文獻[6]標準確立典型RP診斷:(1)有夜盲史;(2)視力逐漸下降;(3)視盤顏色蠟黃或變淡、視網膜骨細胞樣色素沉著及視網膜血管變細;(4)早期視野環形暗點,晚期視野呈向心性縮窄;(5)全視野視網膜電圖(ERG)早期視桿細胞功能降低,可有視錐細胞輕度降低,晚期視錐-視桿細胞功能均降低。參照文獻[7]確立無色素RP(RPSP)診斷:眼底無骨細胞樣色素沉著,其他癥狀和體征同典型RP。參照文獻[8]標準確立Usher綜合征2型(USH2)診斷:符合典型RP眼底表現,同時伴有先天性中重度耳聾,前庭功能正常。參照文獻[9]標準確立CORD診斷:(1)視力逐漸下降;(2)視野存在中心暗點;(3)全視野ERG視錐-視桿細胞功能均降低,并且視錐細胞功能與視桿細胞功能相同或更嚴重受損;(4)多數患者檢眼鏡或光相干斷層掃描(OCT)檢查可見黃斑萎縮。排除Stargardt病、中心性暈輪狀視網膜脈絡膜萎縮、卵黃樣黃斑營養不良、老年性黃斑變性等。
詳細收集患者病史、婚育史及家族史,繪制家系圖。所有受檢者均行最佳矯正視力(BCVA)、裂隙燈顯微鏡、間接檢眼鏡、彩色眼底照相、視野、全視野ERG、OCT檢查。行熒光素眼底血管造影(FFA)檢查43例。患者家庭成員及對照組受檢者均無夜盲病史,眼底檢查未見異常,排除RP和CORD。
抽取所有受檢者外周靜脈血3~5 ml,乙二胺四乙酸抗凝,采用德國QIAGEN公司Qiamp Blood Mini Kit DNA提取試劑盒按照操作規程提取全基因組DNA,濃度≥50 ng/μl,吸光度260/280為1.8~2.0,DNA總量≥6 μl。構建Illumina末端堿基配對文庫,通過美國Invitrogen公司PicoGreen DNA定量試劑盒對構建的DNA文庫進行定量,將總量為1 μg的文庫混合物與美國Agilent公司定制的特異性RNA探針進行雜交,對目前已知的232個IRD致病基因的外顯子區域進行液相捕獲,將獲得的捕獲文庫用Illumina HiSeqTM2000(美國Illumina公司)第二代測序儀進行雙端測序,讀長為100堿基對。讀取的測序結果運用Burrows-Wheeler Aligner軟件與人類基因組對照序列hg19進行比對。通過Genome Analysis Tool Kit工具對測序結果進行校準和完善。單核苷酸多態性(SNP)和小分子的插入或缺失突變通過Atlas SNP和AtlasIndel2軟件進行分析。濾過頻率高于0.5%(隱性變異)或者0.1%(顯性變異)突變位點。分析過程涉及的對照數據庫有千人基因組、dbSNP135、NHLBI外顯子測序數據庫、NIEHS外顯子測序數據庫。在濾過高頻率變異后,運用ANNOVAR軟件對篩選出的突變基因進行蛋白質改變預測,同時剔除同義突變。其后,利用專業版人類基因突變數據庫中已知的232個IRD致病基因位點進行檢索以確定已知的突變位點。通過SIFT、Polyphen2、LRT、GERP++、MutationTaster等不同預測軟件對異義突變位點進行功能性預測,判斷該位點的致病性。所有經過高通量測序檢測出的可能致病突變均通過Sanger測序進行驗證并進行家系共分離驗證。Sanger測序結果利用軟件Sequencher(version 5.0)進行分析,最終確定致病基因突變位點。根據常染色體顯性遺傳(AD)、常染色體隱性遺傳(AR)、X連鎖遺傳(XL)的遺傳規律,分析家族史,確立其遺傳類型。
2 結果
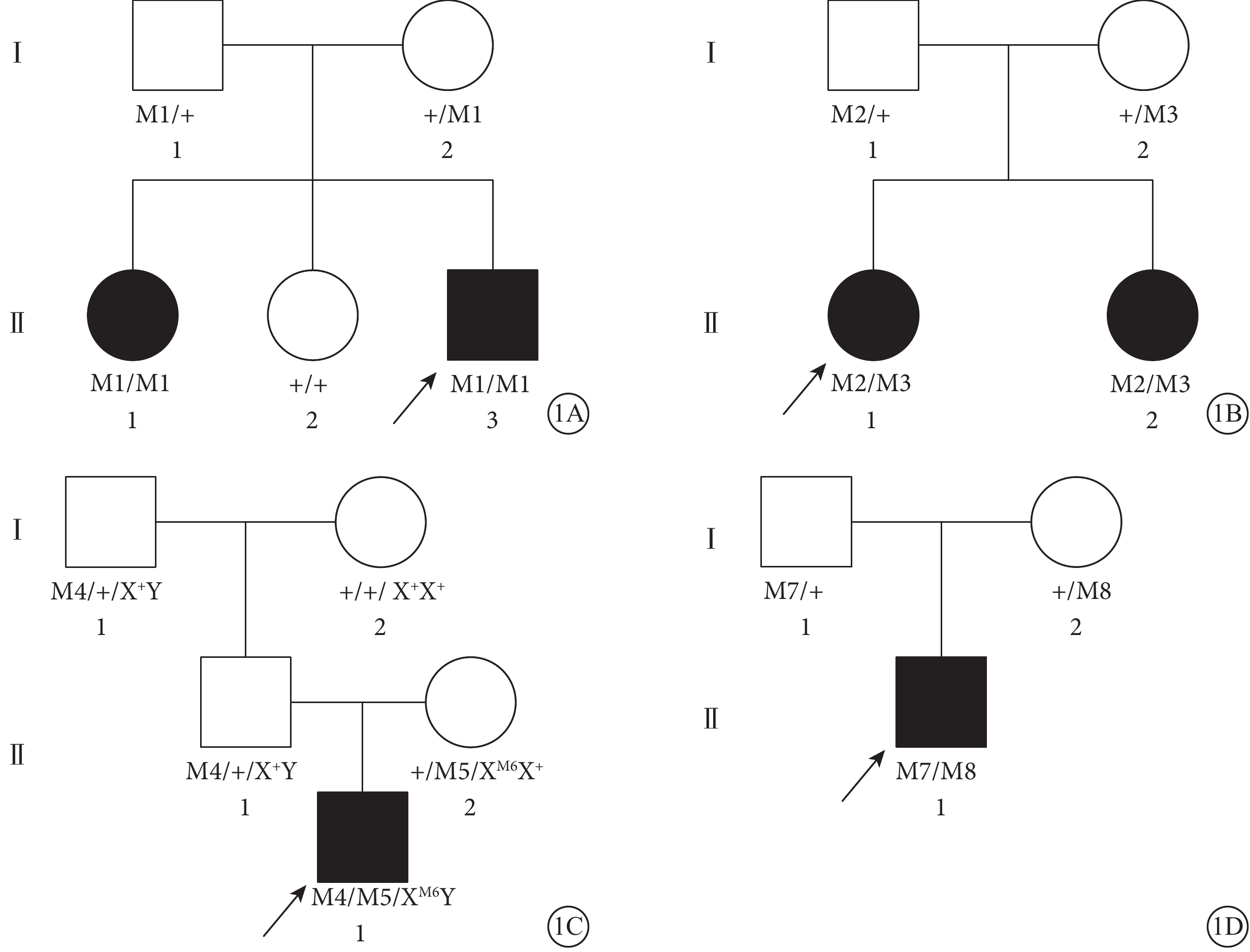
37例RP患者中,13例來自6個家系,其中ADRP 10例(4個家系),ARRP 3例(2個家系);24例為散發RP。6例CORD患者來自4個AR家系(圖1)。
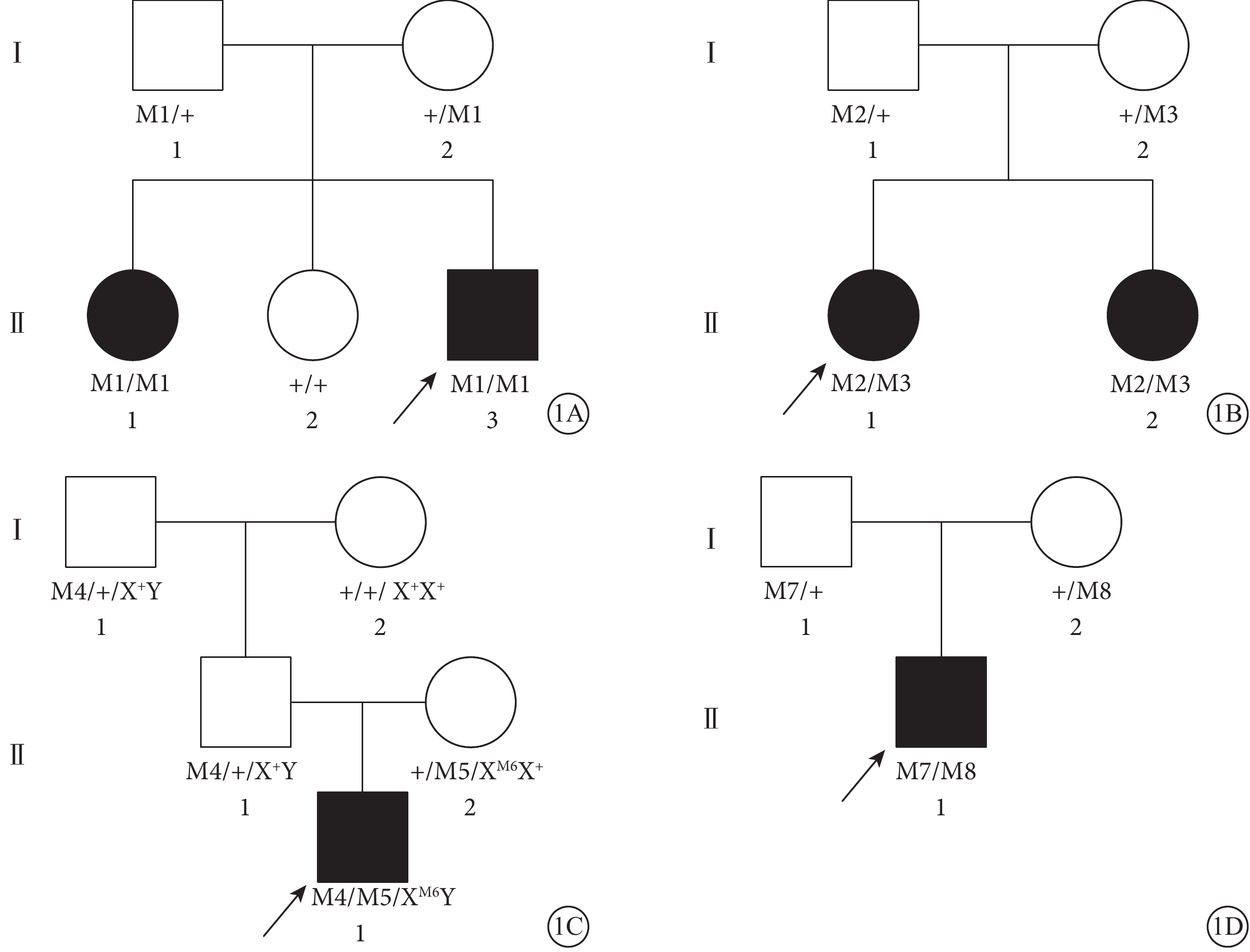 圖1
6例CORD患者家系圖。1A~1D.C01~C04患者家系(家系1~4)。■:男性患者;●:女性患者;↗:先證者;□:正常男性;○:正常女性;M1:p.G410R (c.G1228C)CDATA[M2:p.R1108H (c.3323G>A);M3:p.V521fs (c.1561delG);M4:p.C103R (c.T307C);M5:p.R881Q (c.G2642A);M6:p.C103R (c.A307G);M7:p.R338C (c.C1012T);M8:p.36_42del (c.107_124del)
圖1
6例CORD患者家系圖。1A~1D.C01~C04患者家系(家系1~4)。■:男性患者;●:女性患者;↗:先證者;□:正常男性;○:正常女性;M1:p.G410R (c.G1228C)CDATA[M2:p.R1108H (c.3323G>A);M3:p.V521fs (c.1561delG);M4:p.C103R (c.T307C);M5:p.R881Q (c.G2642A);M6:p.C103R (c.A307G);M7:p.R338C (c.C1012T);M8:p.36_42del (c.107_124del)
43例患者中,檢測出致病性基因突變21例,檢測陽性率48.8%。其中,RP患者15例,檢測出USH2A、RP1、MYO7A、C8orf37、RPGR、SNRNP200、CRX、PRPF31、C2orf71、IMPDH1等10個致病性基因突變;發現突變位點18個,其中12個為新發現突變位點。臨床表型包括典型RP 10 例,RPSP 2例,USH2 3例。CORD患者6例,均檢測到致病性基因突變,其中ABCA4、RIMS1基因突變各2例、CLN3基因突變1例,CRB1和RPGR雙基因突變1例(表1)。對照組受檢者均未檢測到基因突變。
檢測到基因突變的21例患者,BCVA為無光感~0.8。發病年齡4~44歲者17例,自幼者4例;就診時年齡9~59歲。首發癥狀為夜視力下降者12例(RP 10例,RPSP 2例),夜視力下降伴聽力障礙3例(USH2)。6例CORD患者首發癥狀均為視力下降。
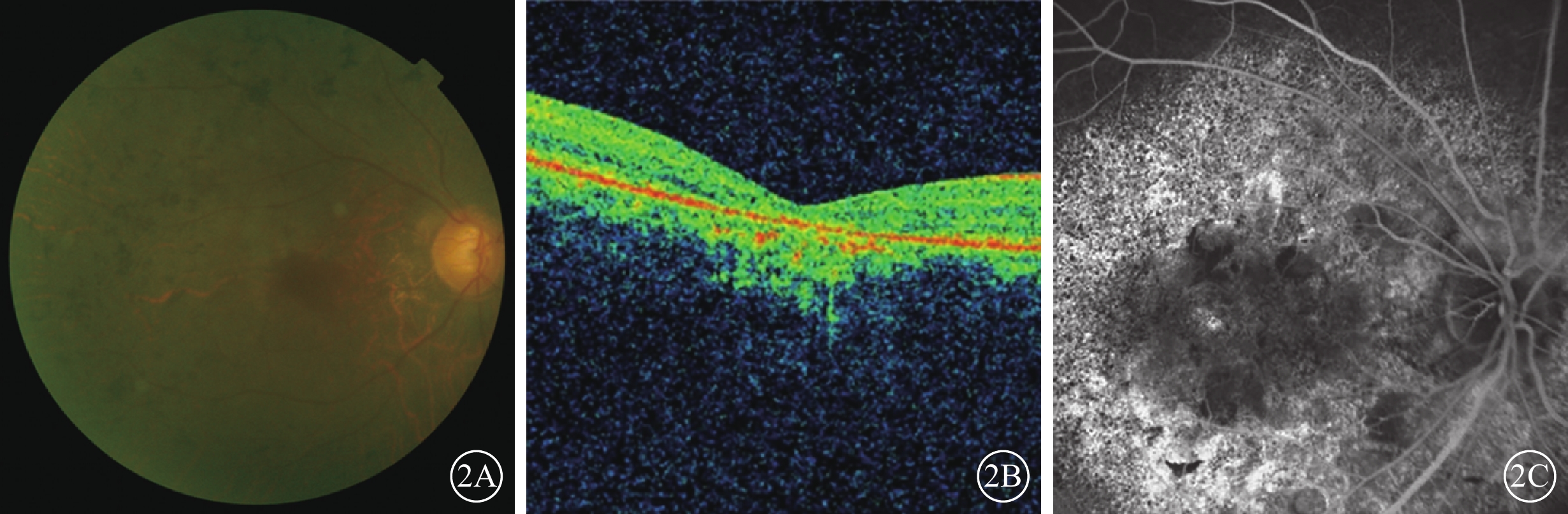
典型RP者7例。患者序號為RP02、RP03、RP06、RP08、RP10、RP11、RP12,致病基因為RP1、C8orf37、RPGR、RPRF31、SNRNP200、CRX、USH2A。突變位點分別為c.1915dupA、c.3937delA、c.172A>T、c.99C>A、c.154G>A、c.97delG、c.C4708T、c.C682T、c.2802T>G、c.14944_14945insCT。其中,c.1915dupA、c.3937delA、c.97delG、c.14944_14945ins CT為新發現的移碼突變;c.C4708T為新發現的錯義突變;c.C682T為新發現的終止突變。患者首診原因均為夜視力下降,BCVA無光感~0.8。視盤顏色明顯變淡或蠟黃,視網膜血管變細,后極部及周邊散在骨細胞樣色素顆粒沉著(圖2A)。OCT檢查,黃斑區萎縮,神經上皮變薄,中心凹變淺,橢圓體帶連續性中斷或消失,視網膜色素上皮(RPE)層變薄(圖2B)。FFA檢查,視網膜呈顆粒樣強熒光伴熒光遮蔽(圖2C)。全視野ERG檢查,暗適應(a波和b波)振幅熄滅3例,重度下降4例;明適應(a波和b波)振幅熄滅4例,重度下降2例,中度下降1例。色覺檢查,紅綠色盲、全色盲分別為4、1例,色覺正常1例;1例患者雙眼視力手動,未行色覺檢查。
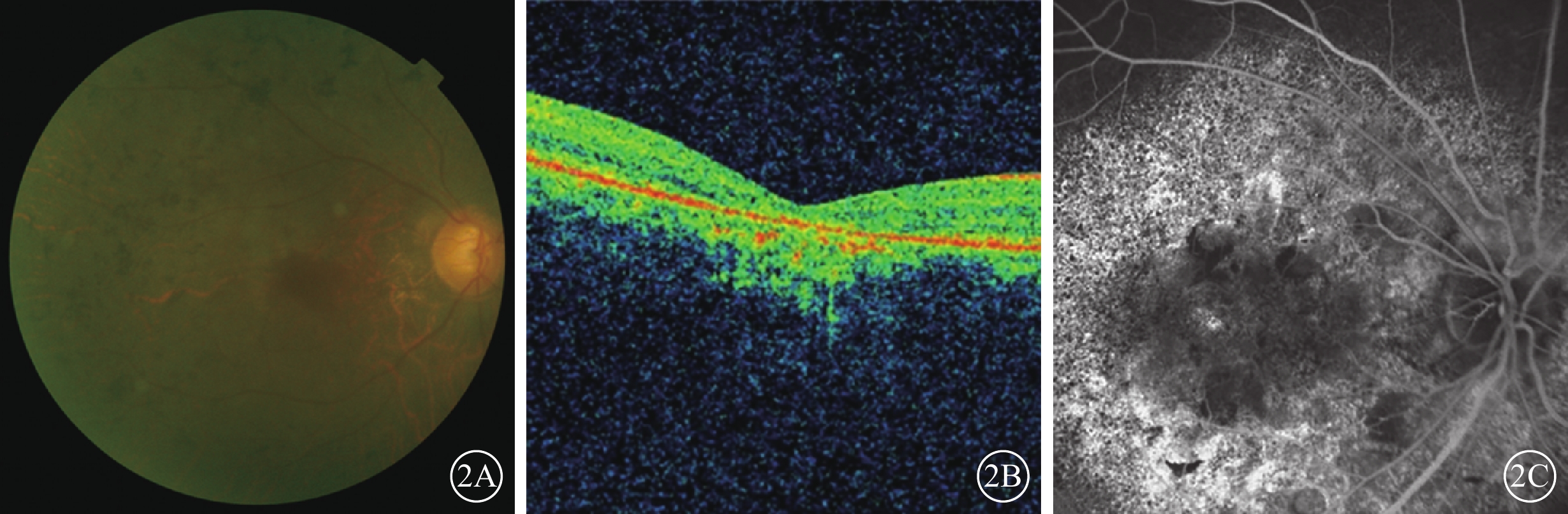 圖2
典型RP患者彩色眼底、OCT、FFA像。2A.彩色眼底像,視網膜骨細胞樣色素沉著;2B.OCT像,黃斑區萎縮,神經上皮層變薄,橢圓體帶消失,RPE層變薄;2C.FFA像,視網膜顆粒狀強熒光
圖2
典型RP患者彩色眼底、OCT、FFA像。2A.彩色眼底像,視網膜骨細胞樣色素沉著;2B.OCT像,黃斑區萎縮,神經上皮層變薄,橢圓體帶消失,RPE層變薄;2C.FFA像,視網膜顆粒狀強熒光
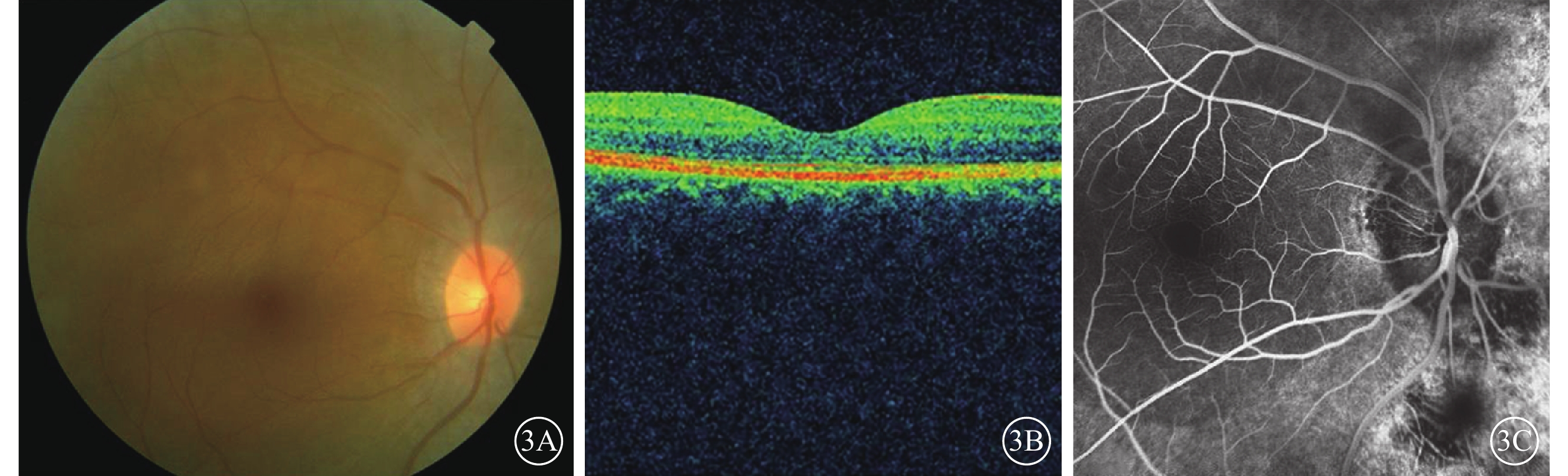
RPSP者2例,患者序號為RP01、RP09。RP01患者致病基因為C2orf71;突變位點為c.3315_3316del、c.1159dupC,均為新發現的移碼突變。患者4歲開始出現夜視力下降;BCVA 0.2。眼底未見視網膜骨細胞樣色素沉著(圖3A);OCT檢查,橢圓體帶連續性中斷(圖3B);FFA檢查,視盤周圍片狀遮蔽熒光,周邊視網膜顆粒狀強熒光(圖3C)。全視野ERG檢查,暗適應、明適應(a波和b波)振幅均為熄滅。色覺檢查,紅綠色盲。序號RP09患者致病基因為IMPDH1;突變位點為c.G1157C,為新發現錯義突變。患者自幼夜盲。BCVA 0.1。眼底未見視網膜骨細胞樣色素沉著;全視野ERG檢查,明適應、暗適應(a波和b波)振幅均重度下降。色覺檢查,全色盲。
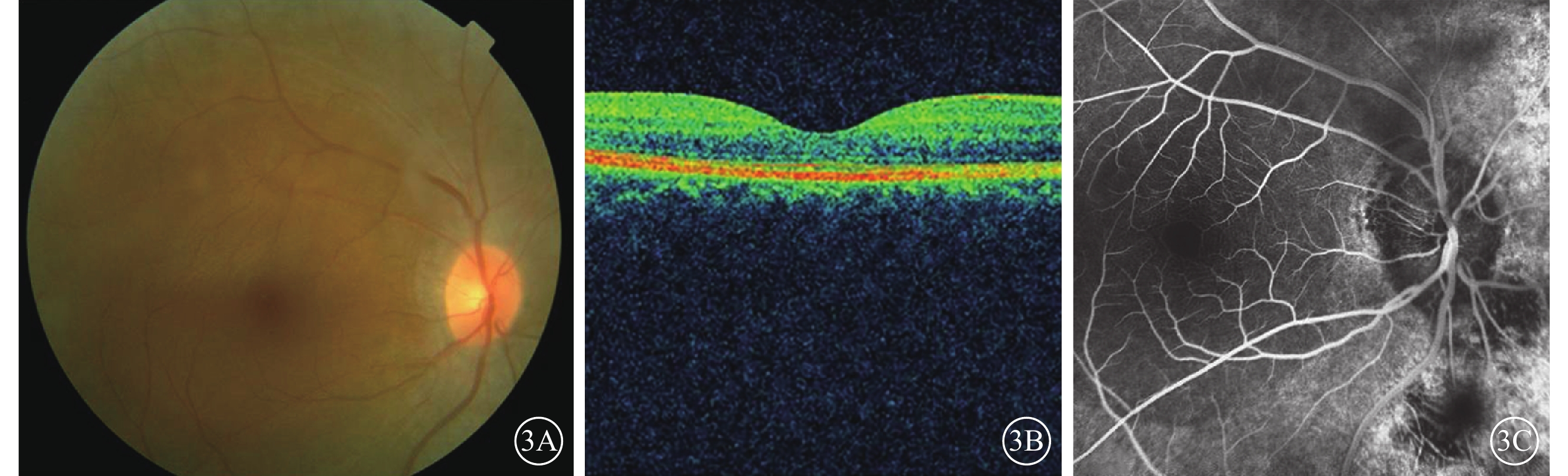 圖3
RPSP患者彩色眼底、OCT、FFA像。3A.彩色眼底像,未見視網膜骨細胞樣色素沉著;3B.OCT像,橢圓體帶連續性中斷;3C.FFA像,視盤周圍片狀遮蔽熒光,周邊視網膜可見顆粒狀強熒光
圖3
RPSP患者彩色眼底、OCT、FFA像。3A.彩色眼底像,未見視網膜骨細胞樣色素沉著;3B.OCT像,橢圓體帶連續性中斷;3C.FFA像,視盤周圍片狀遮蔽熒光,周邊視網膜可見顆粒狀強熒光
USH2者3例,患者序號為RP04、RP05、RP07。致病基因為USH2A、MYO7A。RP04患者致病基因為USH2A;突變位點為c.8559-2A>G、c.5083delA,分別為新發現的剪切突變和移碼突變。其中,剪切突變c.8559-2A>G未引起其所編碼的蛋白質發生改變;移碼突變c.5083delA導致其所編碼的蛋白質(p.S1695fs)發生改變。RP05患者致病基因為USH2A;突變位點為c.10179delG,為新發現的移碼突變。RP07患者致病基因為MYO7A;突變位點為c.2837T>G、c.T5516G,其中c.T5516G為新發現的錯義突變。c.2837T>G所對應的氨基酸改變為MYO7A基因所編碼蛋白的第946號氨基酸從甲硫氨酸突變為精氨酸(p.M946R);c.T5516G所對應的氨基酸改變為MYO7A基因所編碼蛋白的第1839號氨基酸從亮氨酸突變為精氨酸(p.L1839R)。患者自幼有中等程度聽力障礙,前庭功能檢查均正常。BCVA分別為0.6、0.3、手動。眼底視盤顏色淡,視網膜血管變細,周邊骨細胞樣色素沉著。全視野ERG檢查,暗適應(a波和b波)振幅重度下降3例;明適應(a波和b波)振幅重度下降、熄滅分別為2、1例。色覺檢查,紅綠色盲2例;RP07患者未行色覺檢查。
CORD者6例,患者序號為C01、C02、C03、C04。致病基因為RIMS1、ABCA4、RPGR、CRB1、CLN3;突變位點分別為c.G1228C、c.3323G>A、c.1561delG、c.A307G、c.T307C、c.G2642A、c.C1012T、c.107_124del。其中,c.G1228C、c.A307G、c.T307C 、c.C1012T為新發現的錯義突變;c.1561delG、c.107_124del為新發現的移碼突變。
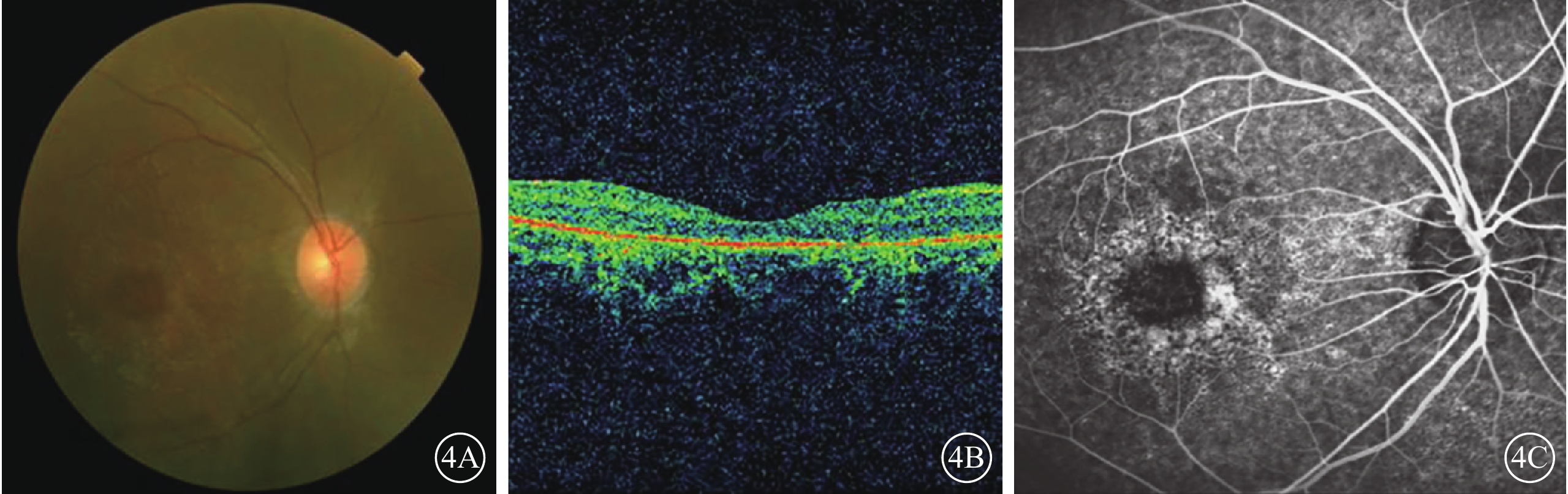
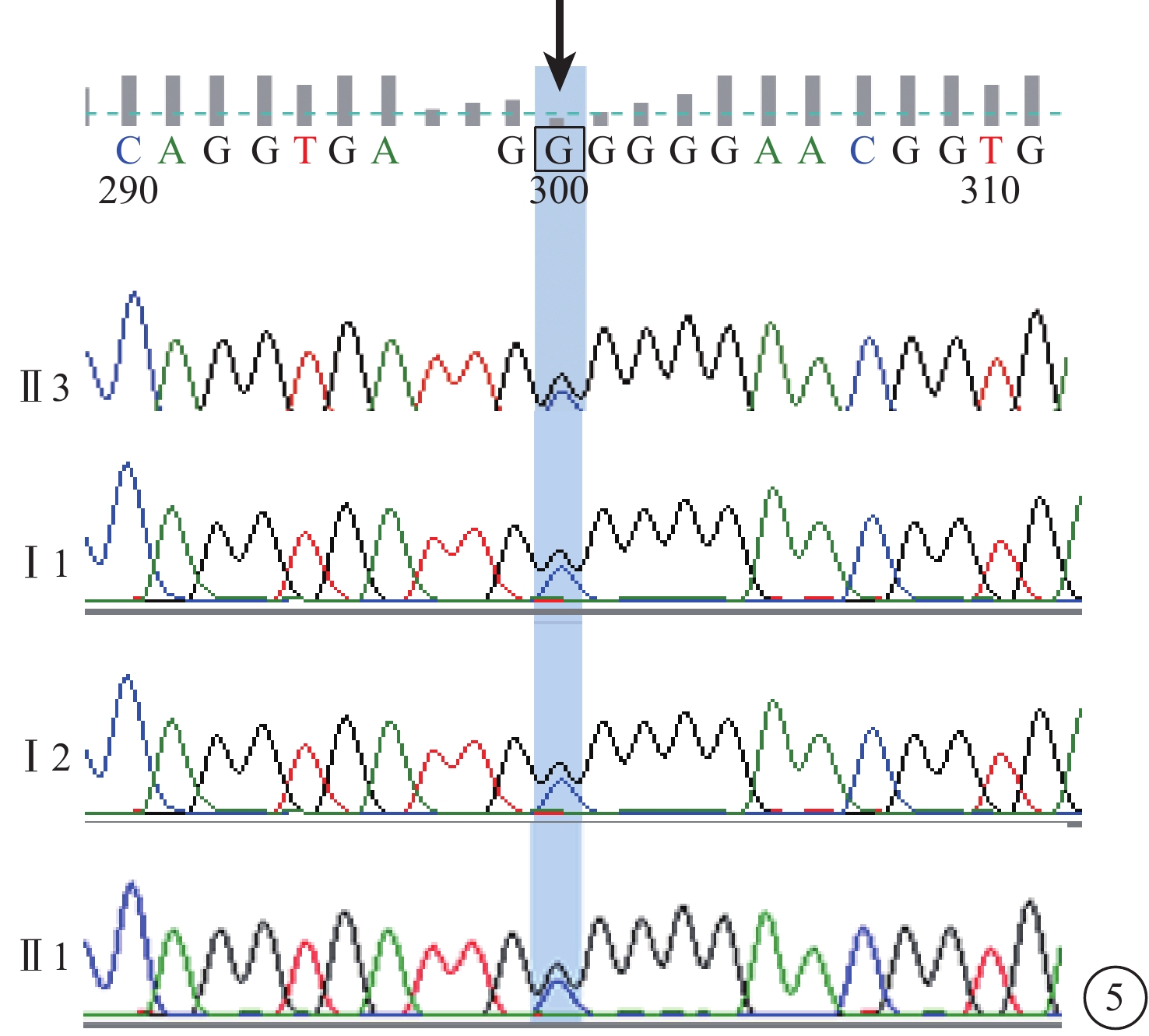
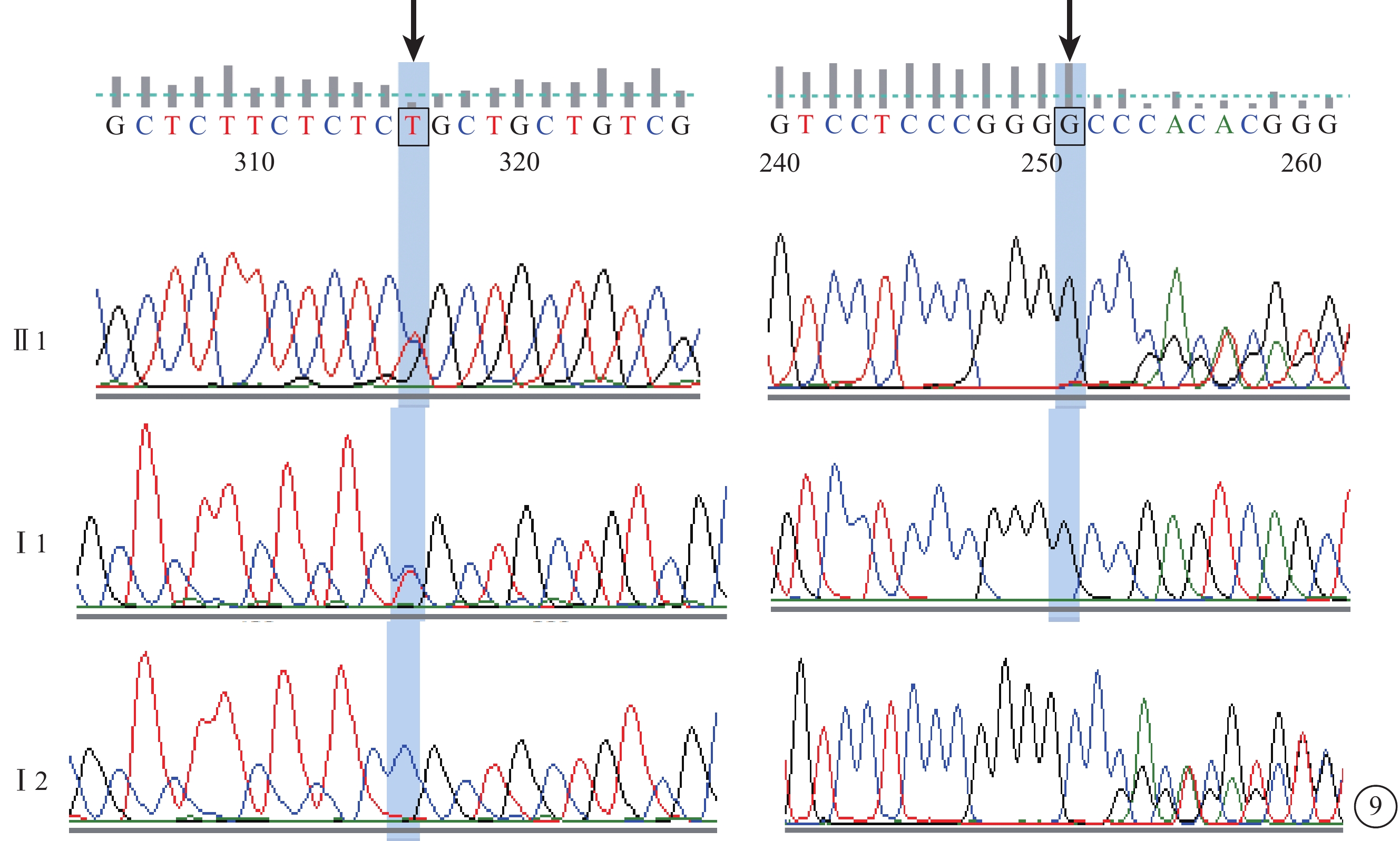
C01患者(家系1先證者Ⅱ3),男,16歲。雙眼視力下降10年;無夜盲史。既往曾診斷為視錐細胞營養不良(COD)。雙眼BCVA均為0.25。視網膜血管纖細(圖4A)。OCT檢查,黃斑萎縮,神經上皮層變薄,橢圓體帶消失(圖4B)。FFA檢查,黃斑區可見圓形強熒光(圖4C)。全視野ERG檢查,視桿細胞反應中度下降,視錐細胞反應重度降低。其同胞姐姐(Ⅱ1)視物不清17年,夜盲2年,曾診斷為RP。眼底視盤顏色淡,視網膜大量骨細胞樣色素沉著。全視野ERG檢查,視錐、視桿細胞反應均呈熄滅型。色覺檢查,均為紅綠色盲。其父母臨床表型均正常。先證者及姐姐的第6染色體RIMS1基因上均檢測到c.G1288C位點純合性錯義突變,父母該位點未見突變(圖5)。根據基因檢測結果并結合臨床表型及病史,修正診斷為CORD。
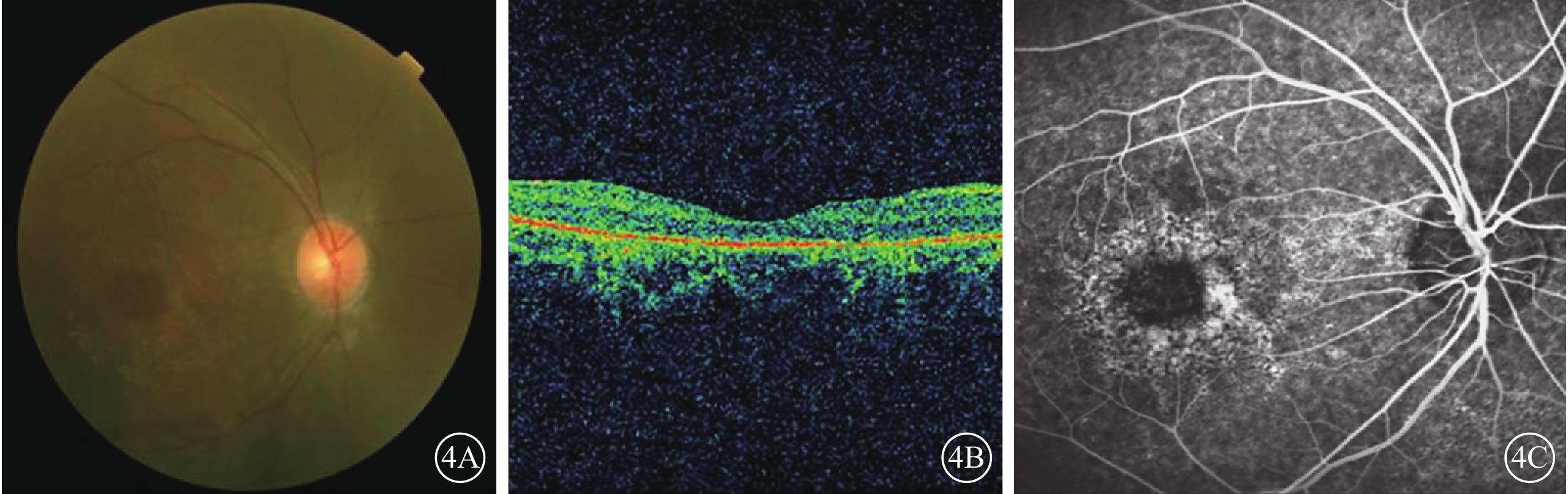 圖4
C01患者彩色眼底、OCT、FFA像。4A.彩色眼底像,視網膜血管纖細;4B.OCT像,黃斑萎縮,神經上皮層變薄,橢圓體帶消失;4C.FFA像,黃斑區類圓形強熒光
圖4
C01患者彩色眼底、OCT、FFA像。4A.彩色眼底像,視網膜血管纖細;4B.OCT像,黃斑萎縮,神經上皮層變薄,橢圓體帶消失;4C.FFA像,黃斑區類圓形強熒光
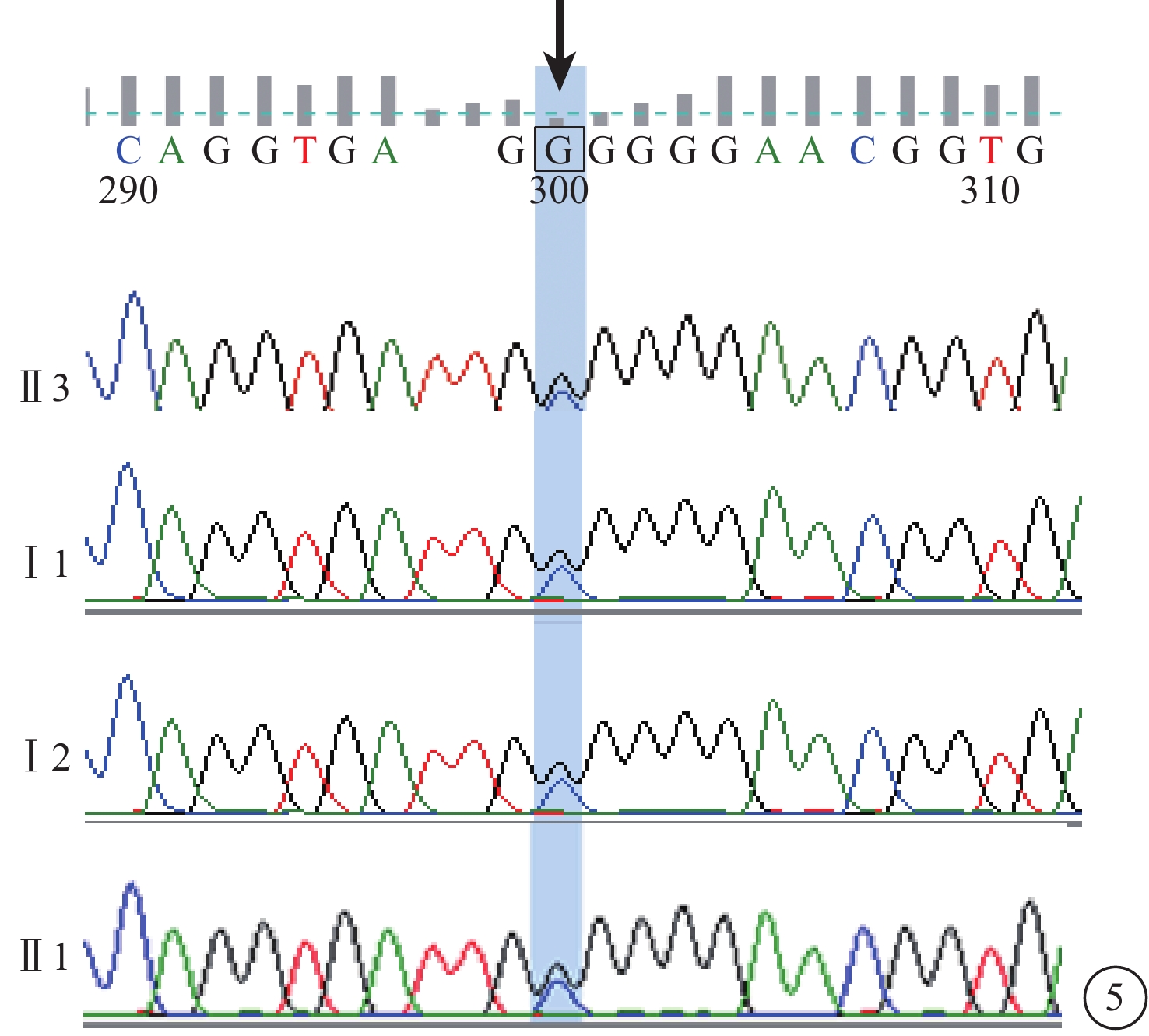 圖5
基因測序圖。先證者(Ⅱ3)及同胞姐姐(Ⅱ1)RIMS1基因純合性錯義突變(c.G1288C)(黑箭);父母(Ⅰ1、Ⅰ2)該基因未見突變(黑箭)
圖5
基因測序圖。先證者(Ⅱ3)及同胞姐姐(Ⅱ1)RIMS1基因純合性錯義突變(c.G1288C)(黑箭);父母(Ⅰ1、Ⅰ2)該基因未見突變(黑箭)
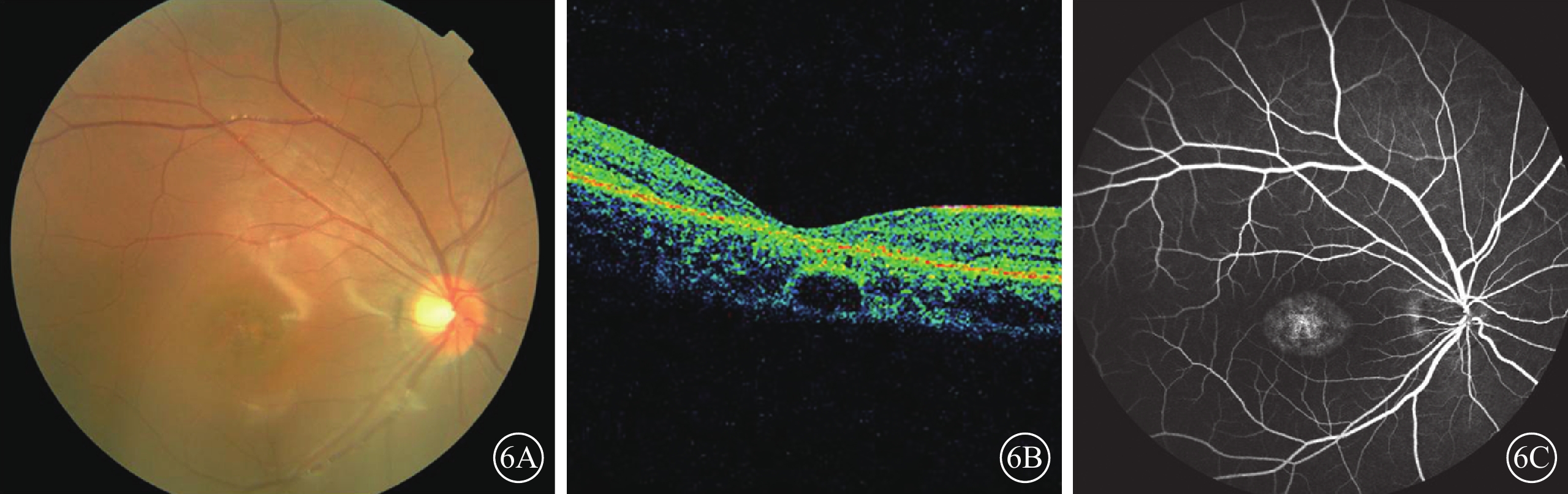
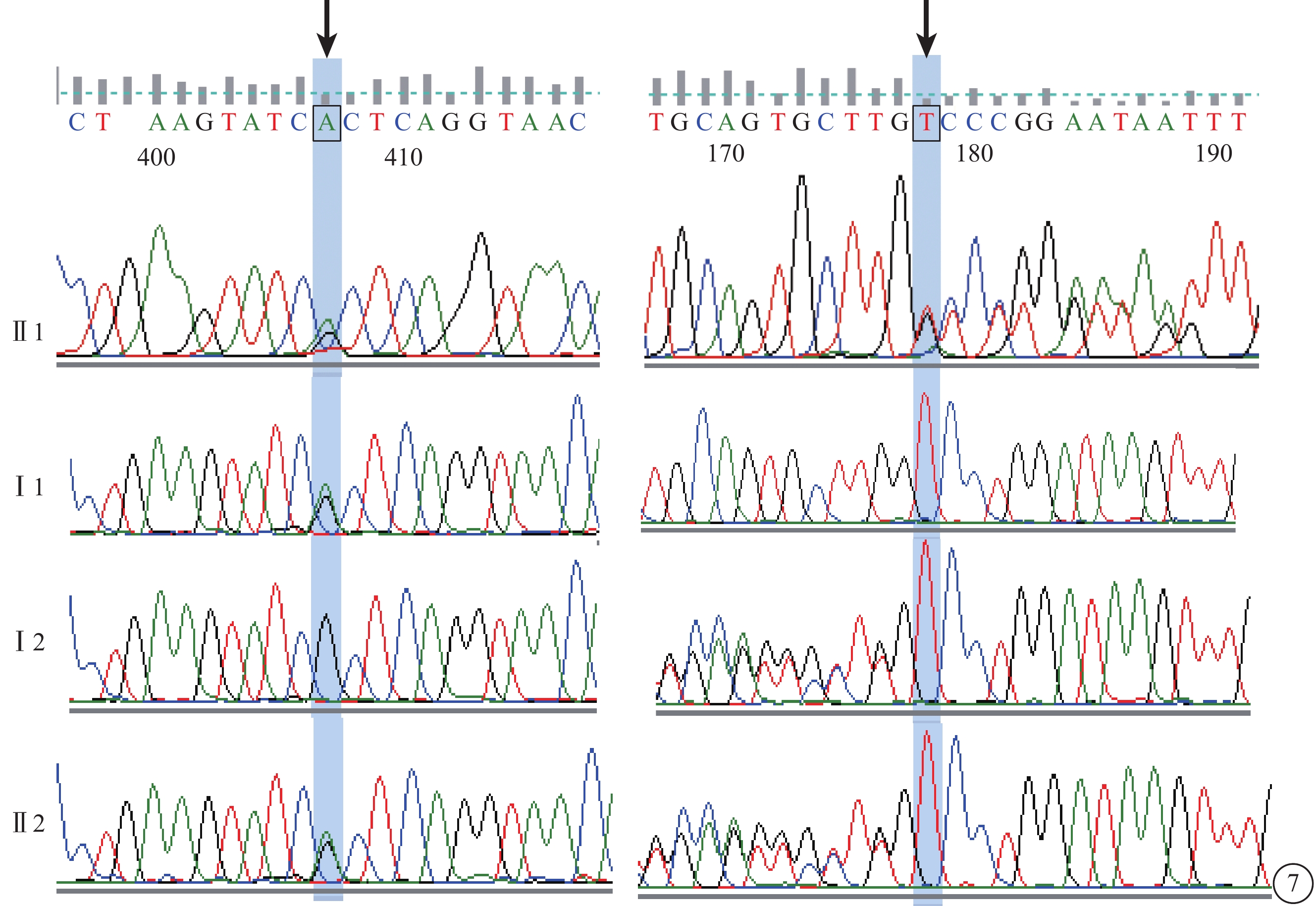
C02患者(家系2先證者Ⅱ1),女,14歲。雙眼視力下降、畏光5年,夜視力下降1年;無夜盲史和眼球震顫。雙眼BCVA分別為 0.2、0.3。雙眼高度近視伴高度散光。眼底黃斑萎縮,色素紊亂,無黃斑斑點沉著(圖6A)。OCT檢查,黃斑萎縮,黃斑中心凹擴大變淺,神經上皮層變薄,橢圓體帶消失,RPE層變薄(圖6B)。FFA檢查,黃斑區可見花環狀強熒光,周邊無顆粒狀強熒光(圖6C)。全視野ERG檢查,暗適應(a波和b波)振幅輕度下降,明適應(a波和b波)振幅重度下降。色覺檢查,正常。其同胞妹妹(Ⅱ4)9歲。雙眼BCVA均為0.5。雙眼高度近視。其母訴暗環境中較同齡兒童行走遲緩。眼底黃斑區色素紊亂,周邊視網膜正常;OCT檢查,黃斑萎縮,黃斑中心凹擴大變淺,神經上皮層變薄,橢圓體帶消失,RPE層變薄;FFA檢查未見明顯異常。全視野ERG檢查,暗適應(a波和b波)振幅中度下降,明適應(a波和b波)振幅重度下降。色覺檢查,正常。其父母(Ⅰ1、Ⅰ2)表型均正常。先證者及同胞妹妹ABCA4基因上均檢測到復合雜合性突變(c.3323G>A、c.1561delG);父母ABCA4基因上分別檢測到錯義突變(c.3323G>A)及移碼突變(c.1561delG)(圖7)。
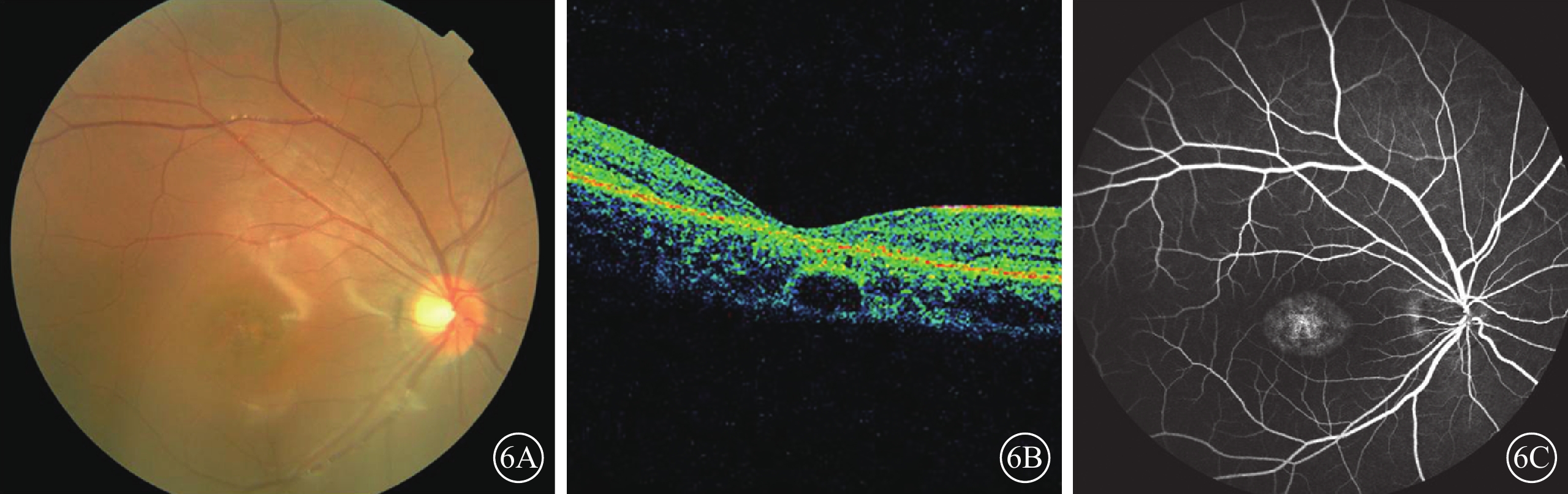 圖6
C02患者彩色眼底、OCT、FFA像。6A.彩色眼底像,黃斑萎縮,周邊視網膜正常,無黃色斑點沉著;6B.OCT像,黃斑萎縮,黃斑中心凹擴大變淺,神經上皮層變薄,橢圓體帶消失,RPE層變薄;6C.FFA像,黃斑區可見花環狀強熒光,周邊無顆粒狀強熒光
圖6
C02患者彩色眼底、OCT、FFA像。6A.彩色眼底像,黃斑萎縮,周邊視網膜正常,無黃色斑點沉著;6B.OCT像,黃斑萎縮,黃斑中心凹擴大變淺,神經上皮層變薄,橢圓體帶消失,RPE層變薄;6C.FFA像,黃斑區可見花環狀強熒光,周邊無顆粒狀強熒光
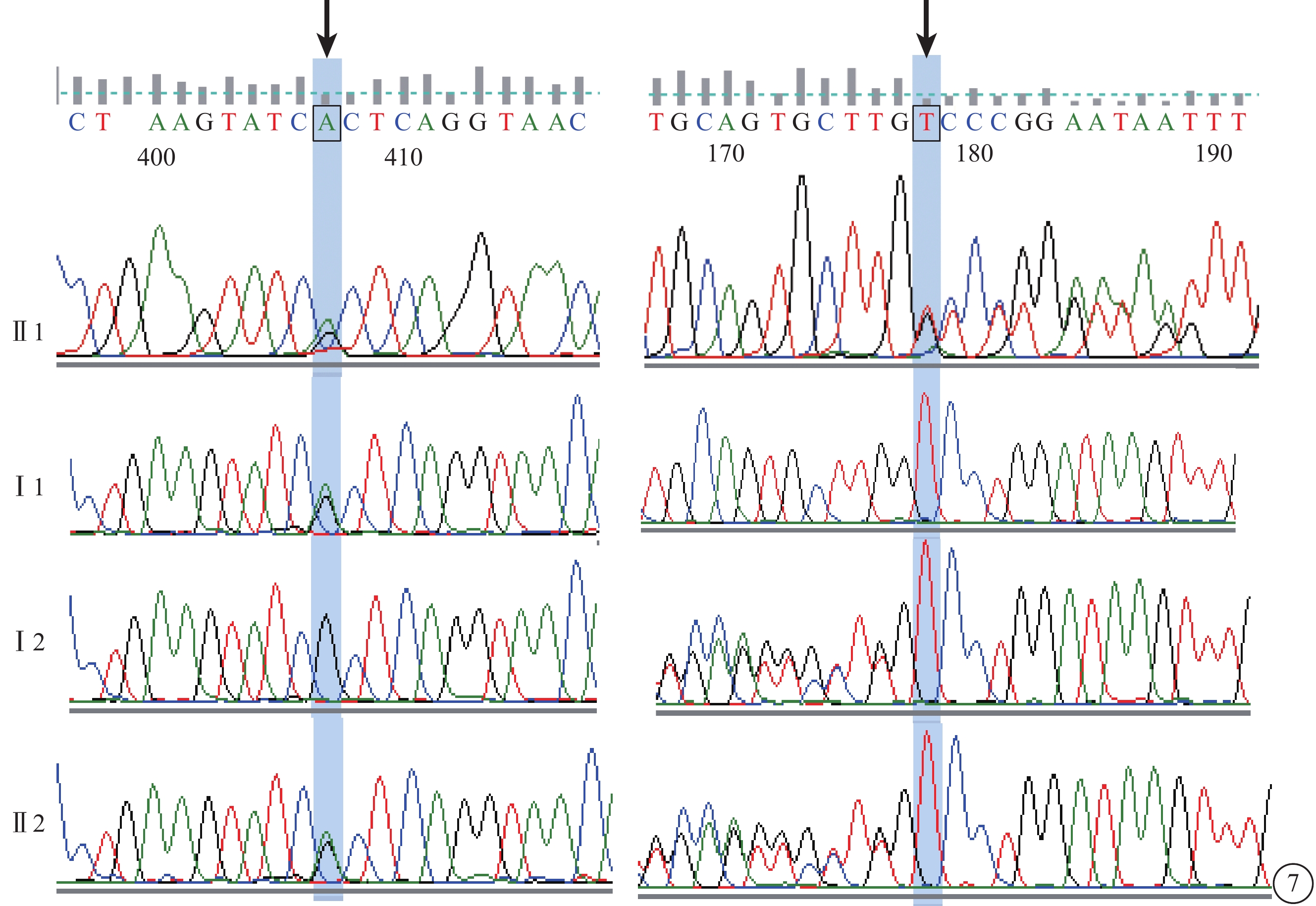 圖7
基因測序圖。先證者(Ⅱ1)及同胞妹妹(Ⅱ2)ABCA4基因復合雜合突變(c.3323G>A、c.1561delG(黑箭);父母(Ⅰ1、Ⅰ2)ABCA4基因錯義突變(c.3323G>A)及移碼突變(c.1561delG)(黑箭)
圖7
基因測序圖。先證者(Ⅱ1)及同胞妹妹(Ⅱ2)ABCA4基因復合雜合突變(c.3323G>A、c.1561delG(黑箭);父母(Ⅰ1、Ⅰ2)ABCA4基因錯義突變(c.3323G>A)及移碼突變(c.1561delG)(黑箭)
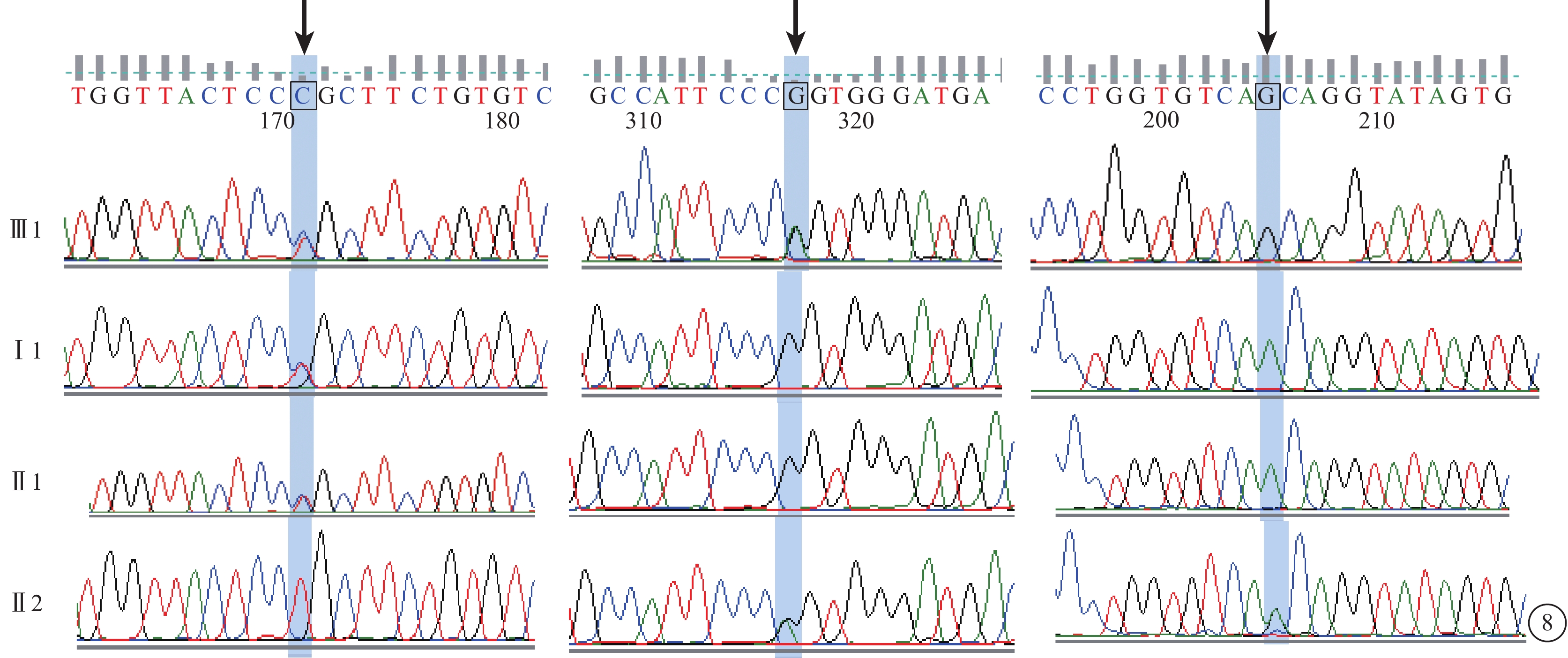
C03患者(家系3先證者Ⅲ1),男,9歲。5歲時發現視力差。雙眼BCVA分別為0.4、0.25。眼底視盤顏色蠟黃、黃斑區少量點狀色素沉著。OCT檢查,黃斑中心凹擴大,橢圓體帶消失,RPE層變薄。FFA檢查,黃斑區類圓形點狀強熒光。全視野ERG檢查,明適應(a波和b波)振幅重度下降,暗適應(a波和b波)振幅輕度降低。視野表現為周邊視野環形縮窄。色覺檢查正常。其爺爺(Ⅰ1)、父母(Ⅱ1、Ⅱ2)表型均正常。先證者同時檢測到RPGR和CRB1雙基因錯義突變(c.A307G、c.T307C、c.G2642A);爺爺、父親CRB1基因上檢測到錯義突變(c.T307C);母親CRB1、RPGR基因上檢測到錯義突變(c.G2642A、c.A307G)(圖8)。
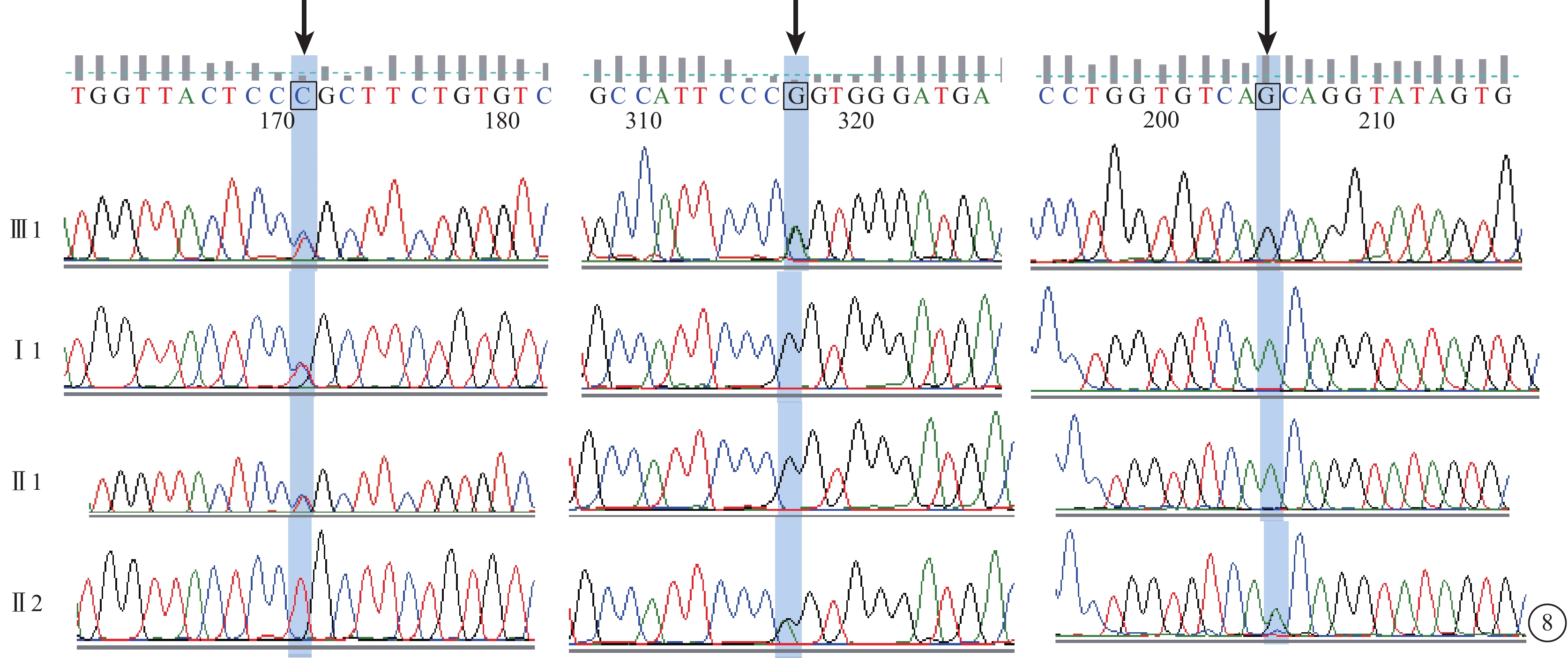 圖8
基因測序圖。先證者(Ⅲ1)RPGR和CRB1雙基因錯義突變(c.A307G、c.T307C、c.G2642A)(黑箭);爺爺(Ⅰ1)、父親(Ⅱ1)CRB1基因錯義突變(c.T307C),母親(Ⅱ2)CRB1、RPGR基因錯義突變(c.G2642A、c.A307G)(黑箭)
圖8
基因測序圖。先證者(Ⅲ1)RPGR和CRB1雙基因錯義突變(c.A307G、c.T307C、c.G2642A)(黑箭);爺爺(Ⅰ1)、父親(Ⅱ1)CRB1基因錯義突變(c.T307C),母親(Ⅱ2)CRB1、RPGR基因錯義突變(c.G2642A、c.A307G)(黑箭)
C04患者(家系4先證者Ⅱ1),男,15歲。自幼視力下降伴夜視力差。雙眼BCVA均為0.15。眼底視盤顏色紅,視網膜血管變細,周邊視網膜無明顯異常。OCT檢查,黃斑區厚度基本正常,橢圓體帶反射信號降低,連續性中斷。FFA檢查,視網膜未見明顯異常。全視野ERG檢查,視桿細胞反應中度下降,視錐細胞反應重度降低。色覺檢查,紅綠色盲。其父母(Ⅰ1、Ⅰ2)均無夜盲史。父親雙眼裸眼視力0.3;色覺檢查,正常;母親雙眼BCVA 0.8,色覺檢查,紅綠色盲。先證者CLN3基因上檢測到雙等位基因錯義突變(c.C1012T)和移碼突變(c.107_124del)。父親CLN3基因的第14號外顯子檢測到新的錯義突變(c.C1012T);母親CLN3基因的第2號外顯子檢測到新的移碼突變(c.107_124del)(圖9)。
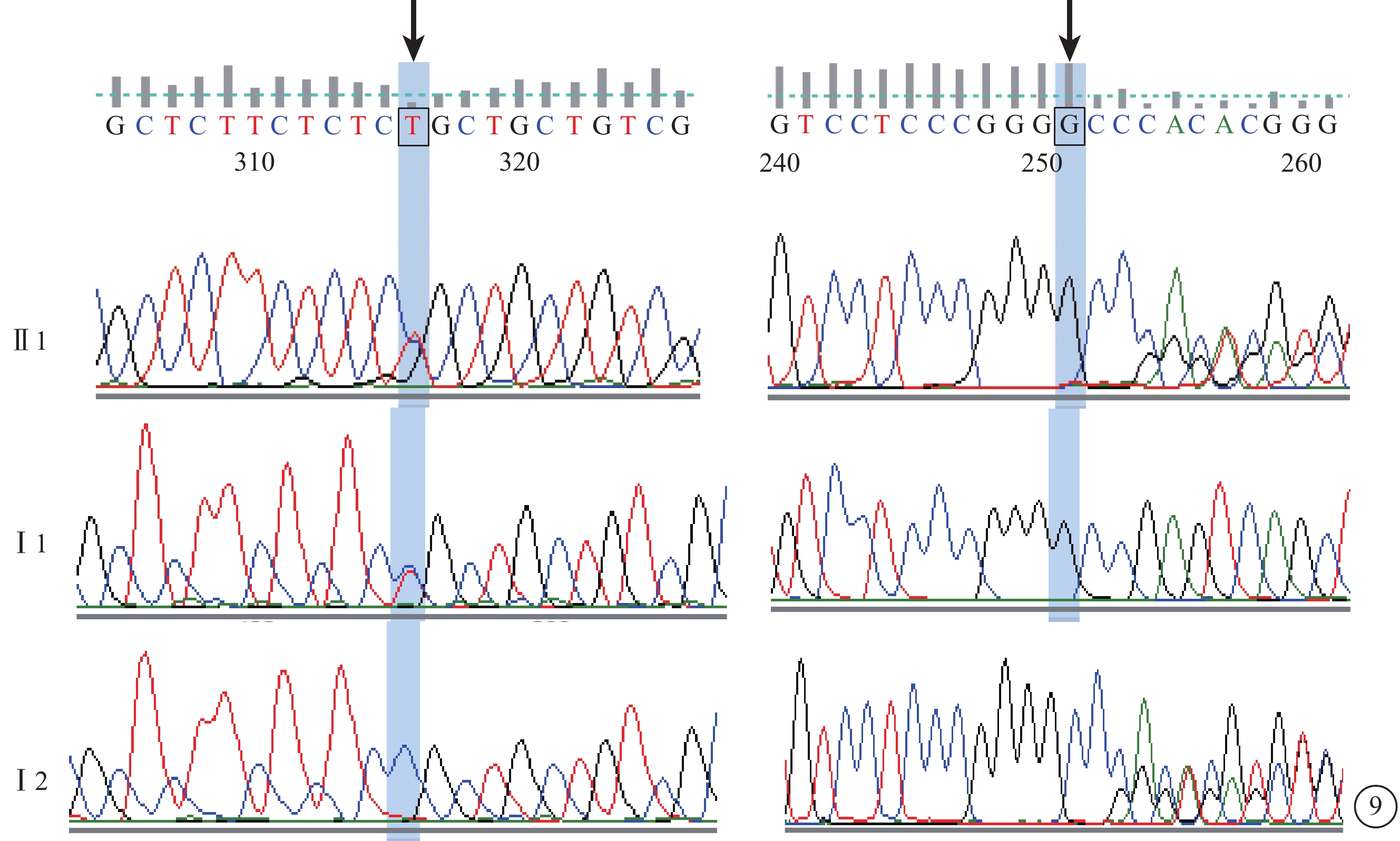 圖9
基因測序圖。先證者(Ⅱ1)CLN3基因雙等位基因錯義突變(c.C1012T)和移碼突變(c.107_124del)(黑箭);父親(Ⅰ1)CLN3基因錯義突變(c.C1012T),母親((Ⅰ2))CLN3基因移碼突變(c.107_124del)(黑箭)
圖9
基因測序圖。先證者(Ⅱ1)CLN3基因雙等位基因錯義突變(c.C1012T)和移碼突變(c.107_124del)(黑箭);父親(Ⅰ1)CLN3基因錯義突變(c.C1012T),母親((Ⅰ2))CLN3基因移碼突變(c.107_124del)(黑箭)
3 討論
本研究應用目標序列捕獲結合第二代測序技術對43例IRD患者進行突變基因篩查,其中21例患者檢測到致病性突變位點,檢測陽性率48.8%。證實該技術可為近50%的RP和CORD患者提供快速且準確的分子診斷。21例患者中,包括15例RP患者和6例CORD患者。15例RP患者中,典型RP 10例、USH2 3例、RPSP 2例,臨床表型多樣化;涉及10個不同致病基因,表現出高度遺傳異質性和臨床異質性。同一致病基因不同突變位點可導致不同的臨床表型,如RP012、RP04患者均檢測到USH2A基因復合雜合性突變,但不同突變位導致RP012患者臨床表型為RP(c.2802T>G、c.14944_14945 insCT),而RP04患者臨床表型為USH2(c.8559-2A>G、c.5083delA)。Xu等[10]在一個中國ARRP家系中檢測到c.2802T> G雜合性錯義突變位點,其臨床表型為典型RP,與本研究中RP012患者臨床表型一致。該位點在多個不同物種中高度保守,c.2802T>G雜合性錯義突變導致第934位氨基酸殘基周圍二級結構的改變,從而干擾蛋白質的正確折疊。Dai等[11]在中國5個USH2家系中檢測到2個家系均存在USH2A基因雜合性剪切突變位點c.8559-2A>G;其臨床表型BCVA光感~0.3,視力損害嚴重,且夜盲開始出現在14~15歲,夜盲發病年齡早。本研究中RP04患者夜盲發病年齡為6歲,雙眼BCVA 0.6。較Dai等[11]研究中患者夜盲發病年齡更早,但BCVA相對較好。其原因可能與RP04患者存在另一個新的移碼突變c.5083delA有關。使用剪接位點預測軟件顯示USH2A基因位點c.8559-2A>G發生雜合性剪切突變后引起外顯子43的剪接受體不能合成,從而使外顯子43不能正常表達,直接或間接導致USH2A翻譯的提前終止或導致超過一個氨基酸殘基的缺失,這可能導致初級蛋白質結構的顯著變化。相同的眼底表現可由不同的致病基因引起。本研究中典型RP患者由RP1、C8orf37、RPGR、RPRF31、SNRNP200、CRX、USH2A等7個不同致病基因突變所致。典型RP患者根據病史、眼底表現診斷不難。RPSP從癥狀和體征上需與先天性靜止性夜盲相鑒別,兩者均有夜盲癥狀且眼底檢查均無骨細胞樣色素沉著,可根據ERG和眼電圖等電生理檢查可明確診斷,同時基因檢測也可為臨床診斷提供可靠依據[12]。
RIMS1基因在腦和視網膜光感受器中表達,定位于帶狀突觸中的突觸前帶,蛋白質產物被認為在突觸傳遞和可塑性中起重要作用,可引起AD-CORD,通常表現為遲發型視錐視桿細胞功能障礙,發病年齡20~50歲,發病年齡越早,預后視力越差[13]。本研究中C01家系中患者在第6染色體RIMS1基因上檢測到c.G1288C位點純合性錯義突變。先證者初診時診斷為COD,實則是CORD病程的第一個階段(早期);而其姐姐診斷為RP,為CORD病程的第二個階段(晚期)。由此可見,CORD在表型上與COD和RP有重疊。CORD在病程的第一階段需與COD鑒別,兩者首先有視錐細胞功能異常, 隨后伴有不同程度的視桿細胞功能異常,COD伴遲發或者輕微的視桿細胞功能障礙,CORD伴顯著的視桿細胞功能障礙。COD在臨床表現為進行性視錐細胞功能障礙伴遲發或者輕微的視桿細胞功能障礙。其臨床特點為首先出現視錐細胞功能障礙且以此為主,視桿細胞受累較晚且輕,很少出現周邊視野缺損,也不會出現夜盲。ERG檢查視錐細胞反應重度下降,視桿細胞反應正常或輕度下降。CORD首先于黃斑區出現視網膜外層的光感受器萎縮,隨年齡增長,病變范圍擴大,可累計周邊部甚至全視網膜;臨床表現為進行性視錐細胞功能障礙伴顯著視桿細胞變性。ERG檢查視錐細胞反應明顯下降或呈熄滅型,視桿細胞下降反應相對輕微。CORD病程的第二個階段需要與RP鑒別。典型RP首發癥狀是夜盲,該癥狀可以獨立存在多年而中心視力完全正常。ERG檢查視錐細胞和視桿細胞反應均重度下降。CORD出現夜盲的時間較晚,嚴重中心視力喪失早于RP。臨床上,如果未行ERG及其他檢查,沒有對家系所有成員完整的臨床表型進行仔細分析,早期一般被診斷COD,晚期易被診斷為RP。
本研究中C02患者及同胞妹妹ABCA4基因上均檢測到復合雜合性突變(c.3323G>A、c.1561delG)。既往研究表明,ABCA4基因既可引起CORD,同時又可以引起RP、Stargardt病等IRD[14-16]。Maugeri等[17]發現ABCA4基因是引起AR-CORD的主要基因。Webster等[18]對374例Stargardt病患者、182例AMD患者及96名正常人ABCA4等位基因變異進行分析,發現1例Stargardt病患者為c.3323G>A位點雜合性錯義突變,該等位基因的患病率低于1%。而C02患者及同胞妹妹臨床表型均為CORD,表明c.3323G>A位點雜合性錯義突變不僅可引起Stargardt病,亦可導致CORD。因此,僅通過基因檢測難以做出準確診斷,需與臨床表型相結合分析,才能確定診斷。C02患者與其同胞妹妹致病基因一樣,但眼底表現存在差異,其妹妹目前黃斑區病變尚不明顯,但隨著病程進展,是否出現與C02患者FFA黃斑區強熒光滲漏一樣的表現,尚有待進一步隨訪觀察。
C03患者檢測到CRB1、RPGR雙基因突變,其臨床表型為CORD。其中在X染色體的RPGR基因上檢測到新發現的純合性錯義突變位點c.A307G,引起第103位的蘇氨酸變異為丙氨酸(p.T103A)。同時在CRB1基因上檢測到復合雜合性突變位點(c.T307C、c.G2642A)。RPGR基因是XLRP中最常見的致病基因,約70%~80%XLRP由RPGR基因突變所致,是臨床表現最為嚴重的RP類型。李自立等[19]在1個6代XLRP家系中發現RPGR基因突變的男性患者表型為嚴重RP。Yang等[20]發現RPGR基因除了可引起XLRP之外還可引起COD,表現為中心視力下降、畏光、色覺障礙三聯征。Thiadens等[21]發現RPGR基因引起的COD患者中,約50%的患者35歲之前視力低于0.5,75%的患者60歲達到法定盲;女性攜帶者則視力不受累。CRB1基因突變引起的RP患者通常發病較早,視力損害程度較重[22]。Papadopoulou Laiou等[23]在5個CRB1基因突變的IRD患者中,發現CRB1基因突變的患者通常在幼年期發病,且表現為自出生起嚴重視力下降,嚴重視野縮小通常在兒童期出現。C03患者發病年齡5歲,雙眼BCVA為0.4、0.25,視野表現為周邊視野環形縮窄,與上述研究結果一致。值得注意的的是本例患者CRB1基因的c.T307C突變來自于其爺爺和父親,而其爺爺和父親臨床表型正常;患者CRB1基因的c.G2642A突變和RPGR基因的c.A307G突變均來自于其母親,其母親臨床表型亦正常。先證者父親和爺爺均攜帶一個突變基因位點,但并未發病;其母親同時攜帶RPGR純合性突變及CRB1雜合性突變位點亦未發病。因而可以推斷RPGR基因純合性突變(c.A307G)和CRB1基因復合雜合性突變(c.T307C、c.G2642A)共同作用導致C03患者CORD的發病。其臨床表型為發病早,病情進展迅速,視力損害程度較重。提示RPGR和CRB1雙基因突變存在潛在的連擊作用。對于CRB1和RPGR同時突變引起CORD的病例,目前尚無文獻報道,究竟是哪個基因突變在患者發病過程中起主導作用目前很難判斷,有待于進一步研究證實。
CLN3基因首先被報道為神經元蠟樣脂褐質沉積癥的致病基因(又名Batten病),定位于第16號染色體,編碼跨膜內體/溶酶體蛋白。Batten病患者由于CLN3基因突變導致跨膜內體/溶酶體蛋白不能夠合成,而引起生物酶的合成受阻。2014年Wang等[24]首次報道了CLN3基因為CORD的新致病基因。Schultz等[25]發現由CLN3基因突變引起的溶酶體沉積病,其患者4~7歲開始出現進行性視力下降,隨后出現運動功能障礙,如帕金森病、癲癇、認知功能障礙。以上研究表明CLN3基因突變不僅可引起神經系統疾病(Batten病),亦可引起IRD。本研究中C04患者在CLN3基因上檢測到新的錯義突變位點(c.C1012T)及新的移碼突變位點(c.107_124del),臨床表型為CORD,C04患者目前無運動系統功能障礙或癲癇發作等異常,隨著病程進展是否出現全身運動系統及神經系統病變,需要隨訪觀察,本研究進一步擴大了CLN3基因突變譜。
根據RetNet網站公布的數據,目前已發現10個不同基因的缺陷可導致AD-COD或AD-CORD的發生,22個基因可導致AR-COD或AR-CORD的發生,2個基因引起XL-COD或XL-CORD。與RP發病有關的基因已經有100多個,其中ABCA4、CRX、C8orf37、CERKL、PROM1、PRPH2、RPGR、SEMA4A等8個基因既可以導致RP又可以導致CORD。由于RP和CORD具有高度遺傳異質性和臨床異質性,兩種疾病在表型之間有一定的相似性和交叉性,鑒于疾病進展和嚴重程度的變異,在不同年齡階段和疾病發展的不同時期,具有明顯的臨床異質性;在致病基因上也存在一定重疊,同一基因不同位點突變可以導致不同疾病,或導致同一疾病但臨床表型不同。因而,對RP和CORD的診斷需依據基因檢測結果,同時要結合細致的臨床表型分析。
視網膜色素變性(RP)和視錐-視桿細胞營養不良(CORD)是臨床上最常見的致盲性遺傳性視網膜疾病(IRD),在早期因缺乏特征性眼部表現時極易漏診和誤診[1-2]。由于兩者具有高度遺傳異質性和臨床異質性[3-5],眾多基因缺陷均可導致上述兩種疾病的發生,尚不能通過基因檢測結果直接進行準確診斷,限制了本病的基因診斷在臨床的廣泛應用。本研究應用目標序列捕獲結合二代測序技術對RP和CORD患者進行致病基因突變檢測,同時結合臨床表型分析,初步探討RP和CORD的診斷和鑒別診斷。現將結果報道如下。
1 對象和方法
2015年9月至2017年4月在寧夏眼科醫院檢查確診的RP患者37例及其三代以內家庭成員84名和CORD患者6例及其三代以內家庭成員11名納入研究。對照組為300名與受檢者無親緣關系的健康成年人。本研究經寧夏回族自治區人民醫院倫理委員會審核批準,遵循赫爾辛基宣言,所有受檢者和未成年患者監護人均簽署知情同意書。
參照文獻[6]標準確立典型RP診斷:(1)有夜盲史;(2)視力逐漸下降;(3)視盤顏色蠟黃或變淡、視網膜骨細胞樣色素沉著及視網膜血管變細;(4)早期視野環形暗點,晚期視野呈向心性縮窄;(5)全視野視網膜電圖(ERG)早期視桿細胞功能降低,可有視錐細胞輕度降低,晚期視錐-視桿細胞功能均降低。參照文獻[7]確立無色素RP(RPSP)診斷:眼底無骨細胞樣色素沉著,其他癥狀和體征同典型RP。參照文獻[8]標準確立Usher綜合征2型(USH2)診斷:符合典型RP眼底表現,同時伴有先天性中重度耳聾,前庭功能正常。參照文獻[9]標準確立CORD診斷:(1)視力逐漸下降;(2)視野存在中心暗點;(3)全視野ERG視錐-視桿細胞功能均降低,并且視錐細胞功能與視桿細胞功能相同或更嚴重受損;(4)多數患者檢眼鏡或光相干斷層掃描(OCT)檢查可見黃斑萎縮。排除Stargardt病、中心性暈輪狀視網膜脈絡膜萎縮、卵黃樣黃斑營養不良、老年性黃斑變性等。
詳細收集患者病史、婚育史及家族史,繪制家系圖。所有受檢者均行最佳矯正視力(BCVA)、裂隙燈顯微鏡、間接檢眼鏡、彩色眼底照相、視野、全視野ERG、OCT檢查。行熒光素眼底血管造影(FFA)檢查43例。患者家庭成員及對照組受檢者均無夜盲病史,眼底檢查未見異常,排除RP和CORD。
抽取所有受檢者外周靜脈血3~5 ml,乙二胺四乙酸抗凝,采用德國QIAGEN公司Qiamp Blood Mini Kit DNA提取試劑盒按照操作規程提取全基因組DNA,濃度≥50 ng/μl,吸光度260/280為1.8~2.0,DNA總量≥6 μl。構建Illumina末端堿基配對文庫,通過美國Invitrogen公司PicoGreen DNA定量試劑盒對構建的DNA文庫進行定量,將總量為1 μg的文庫混合物與美國Agilent公司定制的特異性RNA探針進行雜交,對目前已知的232個IRD致病基因的外顯子區域進行液相捕獲,將獲得的捕獲文庫用Illumina HiSeqTM2000(美國Illumina公司)第二代測序儀進行雙端測序,讀長為100堿基對。讀取的測序結果運用Burrows-Wheeler Aligner軟件與人類基因組對照序列hg19進行比對。通過Genome Analysis Tool Kit工具對測序結果進行校準和完善。單核苷酸多態性(SNP)和小分子的插入或缺失突變通過Atlas SNP和AtlasIndel2軟件進行分析。濾過頻率高于0.5%(隱性變異)或者0.1%(顯性變異)突變位點。分析過程涉及的對照數據庫有千人基因組、dbSNP135、NHLBI外顯子測序數據庫、NIEHS外顯子測序數據庫。在濾過高頻率變異后,運用ANNOVAR軟件對篩選出的突變基因進行蛋白質改變預測,同時剔除同義突變。其后,利用專業版人類基因突變數據庫中已知的232個IRD致病基因位點進行檢索以確定已知的突變位點。通過SIFT、Polyphen2、LRT、GERP++、MutationTaster等不同預測軟件對異義突變位點進行功能性預測,判斷該位點的致病性。所有經過高通量測序檢測出的可能致病突變均通過Sanger測序進行驗證并進行家系共分離驗證。Sanger測序結果利用軟件Sequencher(version 5.0)進行分析,最終確定致病基因突變位點。根據常染色體顯性遺傳(AD)、常染色體隱性遺傳(AR)、X連鎖遺傳(XL)的遺傳規律,分析家族史,確立其遺傳類型。
2 結果
37例RP患者中,13例來自6個家系,其中ADRP 10例(4個家系),ARRP 3例(2個家系);24例為散發RP。6例CORD患者來自4個AR家系(圖1)。
圖1
6例CORD患者家系圖。1A~1D.C01~C04患者家系(家系1~4)。■:男性患者;●:女性患者;↗:先證者;□:正常男性;○:正常女性;M1:p.G410R (c.G1228C)CDATA[M2:p.R1108H (c.3323G>A);M3:p.V521fs (c.1561delG);M4:p.C103R (c.T307C);M5:p.R881Q (c.G2642A);M6:p.C103R (c.A307G);M7:p.R338C (c.C1012T);M8:p.36_42del (c.107_124del)
43例患者中,檢測出致病性基因突變21例,檢測陽性率48.8%。其中,RP患者15例,檢測出USH2A、RP1、MYO7A、C8orf37、RPGR、SNRNP200、CRX、PRPF31、C2orf71、IMPDH1等10個致病性基因突變;發現突變位點18個,其中12個為新發現突變位點。臨床表型包括典型RP 10 例,RPSP 2例,USH2 3例。CORD患者6例,均檢測到致病性基因突變,其中ABCA4、RIMS1基因突變各2例、CLN3基因突變1例,CRB1和RPGR雙基因突變1例(表1)。對照組受檢者均未檢測到基因突變。
檢測到基因突變的21例患者,BCVA為無光感~0.8。發病年齡4~44歲者17例,自幼者4例;就診時年齡9~59歲。首發癥狀為夜視力下降者12例(RP 10例,RPSP 2例),夜視力下降伴聽力障礙3例(USH2)。6例CORD患者首發癥狀均為視力下降。
典型RP者7例。患者序號為RP02、RP03、RP06、RP08、RP10、RP11、RP12,致病基因為RP1、C8orf37、RPGR、RPRF31、SNRNP200、CRX、USH2A。突變位點分別為c.1915dupA、c.3937delA、c.172A>T、c.99C>A、c.154G>A、c.97delG、c.C4708T、c.C682T、c.2802T>G、c.14944_14945insCT。其中,c.1915dupA、c.3937delA、c.97delG、c.14944_14945ins CT為新發現的移碼突變;c.C4708T為新發現的錯義突變;c.C682T為新發現的終止突變。患者首診原因均為夜視力下降,BCVA無光感~0.8。視盤顏色明顯變淡或蠟黃,視網膜血管變細,后極部及周邊散在骨細胞樣色素顆粒沉著(圖2A)。OCT檢查,黃斑區萎縮,神經上皮變薄,中心凹變淺,橢圓體帶連續性中斷或消失,視網膜色素上皮(RPE)層變薄(圖2B)。FFA檢查,視網膜呈顆粒樣強熒光伴熒光遮蔽(圖2C)。全視野ERG檢查,暗適應(a波和b波)振幅熄滅3例,重度下降4例;明適應(a波和b波)振幅熄滅4例,重度下降2例,中度下降1例。色覺檢查,紅綠色盲、全色盲分別為4、1例,色覺正常1例;1例患者雙眼視力手動,未行色覺檢查。
圖2
典型RP患者彩色眼底、OCT、FFA像。2A.彩色眼底像,視網膜骨細胞樣色素沉著;2B.OCT像,黃斑區萎縮,神經上皮層變薄,橢圓體帶消失,RPE層變薄;2C.FFA像,視網膜顆粒狀強熒光
RPSP者2例,患者序號為RP01、RP09。RP01患者致病基因為C2orf71;突變位點為c.3315_3316del、c.1159dupC,均為新發現的移碼突變。患者4歲開始出現夜視力下降;BCVA 0.2。眼底未見視網膜骨細胞樣色素沉著(圖3A);OCT檢查,橢圓體帶連續性中斷(圖3B);FFA檢查,視盤周圍片狀遮蔽熒光,周邊視網膜顆粒狀強熒光(圖3C)。全視野ERG檢查,暗適應、明適應(a波和b波)振幅均為熄滅。色覺檢查,紅綠色盲。序號RP09患者致病基因為IMPDH1;突變位點為c.G1157C,為新發現錯義突變。患者自幼夜盲。BCVA 0.1。眼底未見視網膜骨細胞樣色素沉著;全視野ERG檢查,明適應、暗適應(a波和b波)振幅均重度下降。色覺檢查,全色盲。
圖3
RPSP患者彩色眼底、OCT、FFA像。3A.彩色眼底像,未見視網膜骨細胞樣色素沉著;3B.OCT像,橢圓體帶連續性中斷;3C.FFA像,視盤周圍片狀遮蔽熒光,周邊視網膜可見顆粒狀強熒光
USH2者3例,患者序號為RP04、RP05、RP07。致病基因為USH2A、MYO7A。RP04患者致病基因為USH2A;突變位點為c.8559-2A>G、c.5083delA,分別為新發現的剪切突變和移碼突變。其中,剪切突變c.8559-2A>G未引起其所編碼的蛋白質發生改變;移碼突變c.5083delA導致其所編碼的蛋白質(p.S1695fs)發生改變。RP05患者致病基因為USH2A;突變位點為c.10179delG,為新發現的移碼突變。RP07患者致病基因為MYO7A;突變位點為c.2837T>G、c.T5516G,其中c.T5516G為新發現的錯義突變。c.2837T>G所對應的氨基酸改變為MYO7A基因所編碼蛋白的第946號氨基酸從甲硫氨酸突變為精氨酸(p.M946R);c.T5516G所對應的氨基酸改變為MYO7A基因所編碼蛋白的第1839號氨基酸從亮氨酸突變為精氨酸(p.L1839R)。患者自幼有中等程度聽力障礙,前庭功能檢查均正常。BCVA分別為0.6、0.3、手動。眼底視盤顏色淡,視網膜血管變細,周邊骨細胞樣色素沉著。全視野ERG檢查,暗適應(a波和b波)振幅重度下降3例;明適應(a波和b波)振幅重度下降、熄滅分別為2、1例。色覺檢查,紅綠色盲2例;RP07患者未行色覺檢查。
CORD者6例,患者序號為C01、C02、C03、C04。致病基因為RIMS1、ABCA4、RPGR、CRB1、CLN3;突變位點分別為c.G1228C、c.3323G>A、c.1561delG、c.A307G、c.T307C、c.G2642A、c.C1012T、c.107_124del。其中,c.G1228C、c.A307G、c.T307C 、c.C1012T為新發現的錯義突變;c.1561delG、c.107_124del為新發現的移碼突變。
C01患者(家系1先證者Ⅱ3),男,16歲。雙眼視力下降10年;無夜盲史。既往曾診斷為視錐細胞營養不良(COD)。雙眼BCVA均為0.25。視網膜血管纖細(圖4A)。OCT檢查,黃斑萎縮,神經上皮層變薄,橢圓體帶消失(圖4B)。FFA檢查,黃斑區可見圓形強熒光(圖4C)。全視野ERG檢查,視桿細胞反應中度下降,視錐細胞反應重度降低。其同胞姐姐(Ⅱ1)視物不清17年,夜盲2年,曾診斷為RP。眼底視盤顏色淡,視網膜大量骨細胞樣色素沉著。全視野ERG檢查,視錐、視桿細胞反應均呈熄滅型。色覺檢查,均為紅綠色盲。其父母臨床表型均正常。先證者及姐姐的第6染色體RIMS1基因上均檢測到c.G1288C位點純合性錯義突變,父母該位點未見突變(圖5)。根據基因檢測結果并結合臨床表型及病史,修正診斷為CORD。
圖4
C01患者彩色眼底、OCT、FFA像。4A.彩色眼底像,視網膜血管纖細;4B.OCT像,黃斑萎縮,神經上皮層變薄,橢圓體帶消失;4C.FFA像,黃斑區類圓形強熒光
圖5
基因測序圖。先證者(Ⅱ3)及同胞姐姐(Ⅱ1)RIMS1基因純合性錯義突變(c.G1288C)(黑箭);父母(Ⅰ1、Ⅰ2)該基因未見突變(黑箭)
C02患者(家系2先證者Ⅱ1),女,14歲。雙眼視力下降、畏光5年,夜視力下降1年;無夜盲史和眼球震顫。雙眼BCVA分別為 0.2、0.3。雙眼高度近視伴高度散光。眼底黃斑萎縮,色素紊亂,無黃斑斑點沉著(圖6A)。OCT檢查,黃斑萎縮,黃斑中心凹擴大變淺,神經上皮層變薄,橢圓體帶消失,RPE層變薄(圖6B)。FFA檢查,黃斑區可見花環狀強熒光,周邊無顆粒狀強熒光(圖6C)。全視野ERG檢查,暗適應(a波和b波)振幅輕度下降,明適應(a波和b波)振幅重度下降。色覺檢查,正常。其同胞妹妹(Ⅱ4)9歲。雙眼BCVA均為0.5。雙眼高度近視。其母訴暗環境中較同齡兒童行走遲緩。眼底黃斑區色素紊亂,周邊視網膜正常;OCT檢查,黃斑萎縮,黃斑中心凹擴大變淺,神經上皮層變薄,橢圓體帶消失,RPE層變薄;FFA檢查未見明顯異常。全視野ERG檢查,暗適應(a波和b波)振幅中度下降,明適應(a波和b波)振幅重度下降。色覺檢查,正常。其父母(Ⅰ1、Ⅰ2)表型均正常。先證者及同胞妹妹ABCA4基因上均檢測到復合雜合性突變(c.3323G>A、c.1561delG);父母ABCA4基因上分別檢測到錯義突變(c.3323G>A)及移碼突變(c.1561delG)(圖7)。
圖6
C02患者彩色眼底、OCT、FFA像。6A.彩色眼底像,黃斑萎縮,周邊視網膜正常,無黃色斑點沉著;6B.OCT像,黃斑萎縮,黃斑中心凹擴大變淺,神經上皮層變薄,橢圓體帶消失,RPE層變薄;6C.FFA像,黃斑區可見花環狀強熒光,周邊無顆粒狀強熒光
圖7
基因測序圖。先證者(Ⅱ1)及同胞妹妹(Ⅱ2)ABCA4基因復合雜合突變(c.3323G>A、c.1561delG(黑箭);父母(Ⅰ1、Ⅰ2)ABCA4基因錯義突變(c.3323G>A)及移碼突變(c.1561delG)(黑箭)
C03患者(家系3先證者Ⅲ1),男,9歲。5歲時發現視力差。雙眼BCVA分別為0.4、0.25。眼底視盤顏色蠟黃、黃斑區少量點狀色素沉著。OCT檢查,黃斑中心凹擴大,橢圓體帶消失,RPE層變薄。FFA檢查,黃斑區類圓形點狀強熒光。全視野ERG檢查,明適應(a波和b波)振幅重度下降,暗適應(a波和b波)振幅輕度降低。視野表現為周邊視野環形縮窄。色覺檢查正常。其爺爺(Ⅰ1)、父母(Ⅱ1、Ⅱ2)表型均正常。先證者同時檢測到RPGR和CRB1雙基因錯義突變(c.A307G、c.T307C、c.G2642A);爺爺、父親CRB1基因上檢測到錯義突變(c.T307C);母親CRB1、RPGR基因上檢測到錯義突變(c.G2642A、c.A307G)(圖8)。
圖8
基因測序圖。先證者(Ⅲ1)RPGR和CRB1雙基因錯義突變(c.A307G、c.T307C、c.G2642A)(黑箭);爺爺(Ⅰ1)、父親(Ⅱ1)CRB1基因錯義突變(c.T307C),母親(Ⅱ2)CRB1、RPGR基因錯義突變(c.G2642A、c.A307G)(黑箭)
C04患者(家系4先證者Ⅱ1),男,15歲。自幼視力下降伴夜視力差。雙眼BCVA均為0.15。眼底視盤顏色紅,視網膜血管變細,周邊視網膜無明顯異常。OCT檢查,黃斑區厚度基本正常,橢圓體帶反射信號降低,連續性中斷。FFA檢查,視網膜未見明顯異常。全視野ERG檢查,視桿細胞反應中度下降,視錐細胞反應重度降低。色覺檢查,紅綠色盲。其父母(Ⅰ1、Ⅰ2)均無夜盲史。父親雙眼裸眼視力0.3;色覺檢查,正常;母親雙眼BCVA 0.8,色覺檢查,紅綠色盲。先證者CLN3基因上檢測到雙等位基因錯義突變(c.C1012T)和移碼突變(c.107_124del)。父親CLN3基因的第14號外顯子檢測到新的錯義突變(c.C1012T);母親CLN3基因的第2號外顯子檢測到新的移碼突變(c.107_124del)(圖9)。
圖9
基因測序圖。先證者(Ⅱ1)CLN3基因雙等位基因錯義突變(c.C1012T)和移碼突變(c.107_124del)(黑箭);父親(Ⅰ1)CLN3基因錯義突變(c.C1012T),母親((Ⅰ2))CLN3基因移碼突變(c.107_124del)(黑箭)
3 討論
本研究應用目標序列捕獲結合第二代測序技術對43例IRD患者進行突變基因篩查,其中21例患者檢測到致病性突變位點,檢測陽性率48.8%。證實該技術可為近50%的RP和CORD患者提供快速且準確的分子診斷。21例患者中,包括15例RP患者和6例CORD患者。15例RP患者中,典型RP 10例、USH2 3例、RPSP 2例,臨床表型多樣化;涉及10個不同致病基因,表現出高度遺傳異質性和臨床異質性。同一致病基因不同突變位點可導致不同的臨床表型,如RP012、RP04患者均檢測到USH2A基因復合雜合性突變,但不同突變位導致RP012患者臨床表型為RP(c.2802T>G、c.14944_14945 insCT),而RP04患者臨床表型為USH2(c.8559-2A>G、c.5083delA)。Xu等[10]在一個中國ARRP家系中檢測到c.2802T> G雜合性錯義突變位點,其臨床表型為典型RP,與本研究中RP012患者臨床表型一致。該位點在多個不同物種中高度保守,c.2802T>G雜合性錯義突變導致第934位氨基酸殘基周圍二級結構的改變,從而干擾蛋白質的正確折疊。Dai等[11]在中國5個USH2家系中檢測到2個家系均存在USH2A基因雜合性剪切突變位點c.8559-2A>G;其臨床表型BCVA光感~0.3,視力損害嚴重,且夜盲開始出現在14~15歲,夜盲發病年齡早。本研究中RP04患者夜盲發病年齡為6歲,雙眼BCVA 0.6。較Dai等[11]研究中患者夜盲發病年齡更早,但BCVA相對較好。其原因可能與RP04患者存在另一個新的移碼突變c.5083delA有關。使用剪接位點預測軟件顯示USH2A基因位點c.8559-2A>G發生雜合性剪切突變后引起外顯子43的剪接受體不能合成,從而使外顯子43不能正常表達,直接或間接導致USH2A翻譯的提前終止或導致超過一個氨基酸殘基的缺失,這可能導致初級蛋白質結構的顯著變化。相同的眼底表現可由不同的致病基因引起。本研究中典型RP患者由RP1、C8orf37、RPGR、RPRF31、SNRNP200、CRX、USH2A等7個不同致病基因突變所致。典型RP患者根據病史、眼底表現診斷不難。RPSP從癥狀和體征上需與先天性靜止性夜盲相鑒別,兩者均有夜盲癥狀且眼底檢查均無骨細胞樣色素沉著,可根據ERG和眼電圖等電生理檢查可明確診斷,同時基因檢測也可為臨床診斷提供可靠依據[12]。
RIMS1基因在腦和視網膜光感受器中表達,定位于帶狀突觸中的突觸前帶,蛋白質產物被認為在突觸傳遞和可塑性中起重要作用,可引起AD-CORD,通常表現為遲發型視錐視桿細胞功能障礙,發病年齡20~50歲,發病年齡越早,預后視力越差[13]。本研究中C01家系中患者在第6染色體RIMS1基因上檢測到c.G1288C位點純合性錯義突變。先證者初診時診斷為COD,實則是CORD病程的第一個階段(早期);而其姐姐診斷為RP,為CORD病程的第二個階段(晚期)。由此可見,CORD在表型上與COD和RP有重疊。CORD在病程的第一階段需與COD鑒別,兩者首先有視錐細胞功能異常, 隨后伴有不同程度的視桿細胞功能異常,COD伴遲發或者輕微的視桿細胞功能障礙,CORD伴顯著的視桿細胞功能障礙。COD在臨床表現為進行性視錐細胞功能障礙伴遲發或者輕微的視桿細胞功能障礙。其臨床特點為首先出現視錐細胞功能障礙且以此為主,視桿細胞受累較晚且輕,很少出現周邊視野缺損,也不會出現夜盲。ERG檢查視錐細胞反應重度下降,視桿細胞反應正常或輕度下降。CORD首先于黃斑區出現視網膜外層的光感受器萎縮,隨年齡增長,病變范圍擴大,可累計周邊部甚至全視網膜;臨床表現為進行性視錐細胞功能障礙伴顯著視桿細胞變性。ERG檢查視錐細胞反應明顯下降或呈熄滅型,視桿細胞下降反應相對輕微。CORD病程的第二個階段需要與RP鑒別。典型RP首發癥狀是夜盲,該癥狀可以獨立存在多年而中心視力完全正常。ERG檢查視錐細胞和視桿細胞反應均重度下降。CORD出現夜盲的時間較晚,嚴重中心視力喪失早于RP。臨床上,如果未行ERG及其他檢查,沒有對家系所有成員完整的臨床表型進行仔細分析,早期一般被診斷COD,晚期易被診斷為RP。
本研究中C02患者及同胞妹妹ABCA4基因上均檢測到復合雜合性突變(c.3323G>A、c.1561delG)。既往研究表明,ABCA4基因既可引起CORD,同時又可以引起RP、Stargardt病等IRD[14-16]。Maugeri等[17]發現ABCA4基因是引起AR-CORD的主要基因。Webster等[18]對374例Stargardt病患者、182例AMD患者及96名正常人ABCA4等位基因變異進行分析,發現1例Stargardt病患者為c.3323G>A位點雜合性錯義突變,該等位基因的患病率低于1%。而C02患者及同胞妹妹臨床表型均為CORD,表明c.3323G>A位點雜合性錯義突變不僅可引起Stargardt病,亦可導致CORD。因此,僅通過基因檢測難以做出準確診斷,需與臨床表型相結合分析,才能確定診斷。C02患者與其同胞妹妹致病基因一樣,但眼底表現存在差異,其妹妹目前黃斑區病變尚不明顯,但隨著病程進展,是否出現與C02患者FFA黃斑區強熒光滲漏一樣的表現,尚有待進一步隨訪觀察。
C03患者檢測到CRB1、RPGR雙基因突變,其臨床表型為CORD。其中在X染色體的RPGR基因上檢測到新發現的純合性錯義突變位點c.A307G,引起第103位的蘇氨酸變異為丙氨酸(p.T103A)。同時在CRB1基因上檢測到復合雜合性突變位點(c.T307C、c.G2642A)。RPGR基因是XLRP中最常見的致病基因,約70%~80%XLRP由RPGR基因突變所致,是臨床表現最為嚴重的RP類型。李自立等[19]在1個6代XLRP家系中發現RPGR基因突變的男性患者表型為嚴重RP。Yang等[20]發現RPGR基因除了可引起XLRP之外還可引起COD,表現為中心視力下降、畏光、色覺障礙三聯征。Thiadens等[21]發現RPGR基因引起的COD患者中,約50%的患者35歲之前視力低于0.5,75%的患者60歲達到法定盲;女性攜帶者則視力不受累。CRB1基因突變引起的RP患者通常發病較早,視力損害程度較重[22]。Papadopoulou Laiou等[23]在5個CRB1基因突變的IRD患者中,發現CRB1基因突變的患者通常在幼年期發病,且表現為自出生起嚴重視力下降,嚴重視野縮小通常在兒童期出現。C03患者發病年齡5歲,雙眼BCVA為0.4、0.25,視野表現為周邊視野環形縮窄,與上述研究結果一致。值得注意的的是本例患者CRB1基因的c.T307C突變來自于其爺爺和父親,而其爺爺和父親臨床表型正常;患者CRB1基因的c.G2642A突變和RPGR基因的c.A307G突變均來自于其母親,其母親臨床表型亦正常。先證者父親和爺爺均攜帶一個突變基因位點,但并未發病;其母親同時攜帶RPGR純合性突變及CRB1雜合性突變位點亦未發病。因而可以推斷RPGR基因純合性突變(c.A307G)和CRB1基因復合雜合性突變(c.T307C、c.G2642A)共同作用導致C03患者CORD的發病。其臨床表型為發病早,病情進展迅速,視力損害程度較重。提示RPGR和CRB1雙基因突變存在潛在的連擊作用。對于CRB1和RPGR同時突變引起CORD的病例,目前尚無文獻報道,究竟是哪個基因突變在患者發病過程中起主導作用目前很難判斷,有待于進一步研究證實。
CLN3基因首先被報道為神經元蠟樣脂褐質沉積癥的致病基因(又名Batten病),定位于第16號染色體,編碼跨膜內體/溶酶體蛋白。Batten病患者由于CLN3基因突變導致跨膜內體/溶酶體蛋白不能夠合成,而引起生物酶的合成受阻。2014年Wang等[24]首次報道了CLN3基因為CORD的新致病基因。Schultz等[25]發現由CLN3基因突變引起的溶酶體沉積病,其患者4~7歲開始出現進行性視力下降,隨后出現運動功能障礙,如帕金森病、癲癇、認知功能障礙。以上研究表明CLN3基因突變不僅可引起神經系統疾病(Batten病),亦可引起IRD。本研究中C04患者在CLN3基因上檢測到新的錯義突變位點(c.C1012T)及新的移碼突變位點(c.107_124del),臨床表型為CORD,C04患者目前無運動系統功能障礙或癲癇發作等異常,隨著病程進展是否出現全身運動系統及神經系統病變,需要隨訪觀察,本研究進一步擴大了CLN3基因突變譜。
根據RetNet網站公布的數據,目前已發現10個不同基因的缺陷可導致AD-COD或AD-CORD的發生,22個基因可導致AR-COD或AR-CORD的發生,2個基因引起XL-COD或XL-CORD。與RP發病有關的基因已經有100多個,其中ABCA4、CRX、C8orf37、CERKL、PROM1、PRPH2、RPGR、SEMA4A等8個基因既可以導致RP又可以導致CORD。由于RP和CORD具有高度遺傳異質性和臨床異質性,兩種疾病在表型之間有一定的相似性和交叉性,鑒于疾病進展和嚴重程度的變異,在不同年齡階段和疾病發展的不同時期,具有明顯的臨床異質性;在致病基因上也存在一定重疊,同一基因不同位點突變可以導致不同疾病,或導致同一疾病但臨床表型不同。因而,對RP和CORD的診斷需依據基因檢測結果,同時要結合細致的臨床表型分析。