引用本文: 田敏, 吳進川, 何薇, 余曦, 呂紅彬. 叔丁基對苯二酚對高糖培養視網膜Müller細胞核因子E2相關因子2、血紅素氧合酶1和磷脂酰肌醇-3激酶表達的影響. 中華眼底病雜志, 2018, 34(4): 382-387. doi: 10.3760/cma.j.issn.1005-1015.2018.04.015 復制
糖尿病視網膜病變(DR)發病機制包括氧化應激、細胞凋亡等多方面[1]。核因子E2相關因子2(Nrf2)信號通路是迄今為止發現的最為重要的內源性抗氧化應激通路,其可轉入細胞核內與抗氧化反應元件(ARE)結合并啟動血紅素氧合酶1(HO-1)等下游抗氧化因子轉錄,減輕氧化應激損傷[2]。磷脂酰肌醇-3激酶(PI3K)/蛋白激酶B(Akt)通路在DR中發揮重要作用[3],是細胞存活途徑的主要調節因子[4],其激活可以導致抗凋亡成員B淋巴細胞瘤-2(Bcl-2)增加和促凋亡蛋白Bax的水平降低[5-7]。叔丁基對苯二酚(tBHQ)為Ⅱ相酶誘導劑,是Nrf2/ARE信號通路的強誘導劑。并且也有研究發現tBHQ可以刺激PI3K依賴性Akt磷酸化,上調Nrf2介導的抗氧化應答元件的活性[8, 9]。我們前期研究發現,tBHQ能通過激活Nrf2/ARE信號通路,減輕2型糖尿病大鼠模型視網膜氧化應激損傷,減少視網膜血管增生,抑制視網膜細胞凋亡[10-13]。為此,本研究應用tBHQ預處理高糖環境下大鼠視網膜Müller細胞,觀察tBHQ對高糖環境下大鼠視網膜Müller細胞中Nrf2、HO-1、PI3K、Bcl-2、Bax表達的影響,初步探討Ⅱ相酶誘導劑tBHQ對大鼠視網膜Müller細胞的保護作用及相關機制。現將結果報道如下。
1 材料和方法
Sprague-Dawley大鼠視網膜Müller細胞由武漢生命科技股份有限公司提供。兔抗大鼠谷氨酰胺合成酶(GS)單克隆抗體、兔抗Nrf2多克隆抗體、兔抗Bcl-2多克隆抗體、兔抗甘油醛-3-磷酸脫氫酶(GAPDH)多克隆抗體(英國Abcam公司);異硫氰酸熒光素(FITC)偶聯的山羊抗兔二抗、RIPA總蛋白裂解液、BCA蛋白質濃度測定試劑盒、4’,6-二脒基-2-苯基吲哚(DAPI)(武漢阿斯本生物技術有限公司);兔抗HO-1多克隆抗體(美國Affbiotech公司);兔抗PI3K多克隆抗體(美國CST公司),兔抗Bax多克隆抗體(武漢三鷹生物技術有限公司);Trizol試劑(美國Invitrogen公司),PrimeScriptTMRT試劑盒(日本TaKaRa公司);錨定蛋白(Annexin V)-FITC細胞凋亡檢測試劑盒(天津三箭生物技術有限公司)。
參照文獻[14]的方法培養并鑒定Müller細胞,取第2~4代細胞進行實驗。按照培養液的葡萄糖濃度分為正常糖組(N組,5.5 mmol/L葡萄糖),高糖組(HG組,45.0 mmol/L葡萄糖)及tBHQ干預組(HG+tBHQ組)。參照文獻[15]方法在HG+tBHQ組視網膜Müller細胞培養48 h后,加入tBHQ 20 μmol/L干預處理48 h。
免疫熒光染色法鑒定Müller細胞,GS抗體和DAPI染色觀察Müller細胞形態。采用胰蛋白酶消化第2代對數生長期的Müller細胞,將Müller細胞用4%多聚甲醛固定在蓋玻片上15 min,磷酸鹽緩沖液(PBS)洗滌5 min。室溫下封閉工作流體處理蓋玻片10 min。PBS洗滌5 min,山羊血清室溫下封閉15 min。細胞與GS抗體在4℃溫育過夜。PBS洗滌蓋玻片,與FITC偶聯的山羊抗兔二抗在室溫下孵育50 min。PBS洗滌后,DAPI染色5 min,PBS洗滌3次,滴加適量的抗熒光淬滅劑于Müller細胞上,熒光顯微鏡下觀察。
蛋白免疫印跡法(Western blot)檢測各組Müller細胞中Nrf2、HO-1、PI3K、Bcl-2、Bax蛋白的表達。胰蛋白酶消化第2代對數生長期的Müller細胞,低速離心收集細胞,裂解液沖洗抽提蛋白,4 ℃ 13 000 g離心5 min,收集上清,每孔40 μg蛋白加上樣緩沖液后煮沸5 min,12%十二烷基硫酸鈉聚丙烯酞胺凝膠電泳,轉至聚偏氟乙烯膜,封閉。加入兔抗鼠Nrf2一抗(1∶1000),HO-1一抗(1∶2000),PI3K一抗(1∶3000),Bcl-2一抗(1∶1000)和Bax一抗(1∶1000)4 ℃孵育過夜,洗滌后加羊抗兔二抗(1∶3000),室溫孵育30 min,洗滌后加入新鮮配制的增強化學發光混合溶液到膜的蛋白面側,暗室中曝光。膠片進行掃描存檔,AlphaEaseFC軟件處理系統分析目標條帶的吸光度[A,舊稱光密度(OD)]值。以Nrf2、HO-1、PI3K、Bcl-2、Bax和GAPDH的比值作為各蛋白的相對表達量。
參照文獻[16]方法,實時熒光定量聚合酶鏈(qRT-PCR)檢測各組Müller細胞中Nrf2、HO-1、PI3K、Bcl-2、Bax mRNA的表達。Nrf2、HO-1、PI3K、Bcl-2、Bax、GAPDH引物長度分別為294、290、262、214、148、210堿基對(bp)。72~95 ℃,每上升0.5 ℃取1次熒光值,最后生成溶解曲線,所有的樣本均重復檢測3次,最后計算出Ct值。各因子的表達量采用△△Ct方法進行計算分析,即通過計算目的基因相對于參照因子的倍數來比較mRNA的表達差異,管家基因GAPDH作為內參基因。△△Ct=(待測樣品目的基因的Ct平均值?待測樣本內參基因的Ct平均值)?(對照樣品目的基因的Ct平均值)。目的基因的相對拷貝量F=2?△△Ct。
參照文獻[17]方法,流式細胞儀檢測各組Müller細胞的凋亡率。采用15 mW氬離子激光,激發光波長為488 nm,發射波長530 nm,CEL LQuest軟件自動獲取1×104個細胞樣品。流式細胞儀定量分析,計算Annexin V-FITC及碘化丙啶(PI)雙染陽性細胞百分比含量。
SPSS17.0軟件行統計學處理。結果以均數±標準差(
 ±s)表示。相同指標多組間比較行單因素方差分析,組間兩兩比較采用最小顯著差法檢驗。P<0.05為差異有統計學意義。
±s)表示。相同指標多組間比較行單因素方差分析,組間兩兩比較采用最小顯著差法檢驗。P<0.05為差異有統計學意義。
2 結果
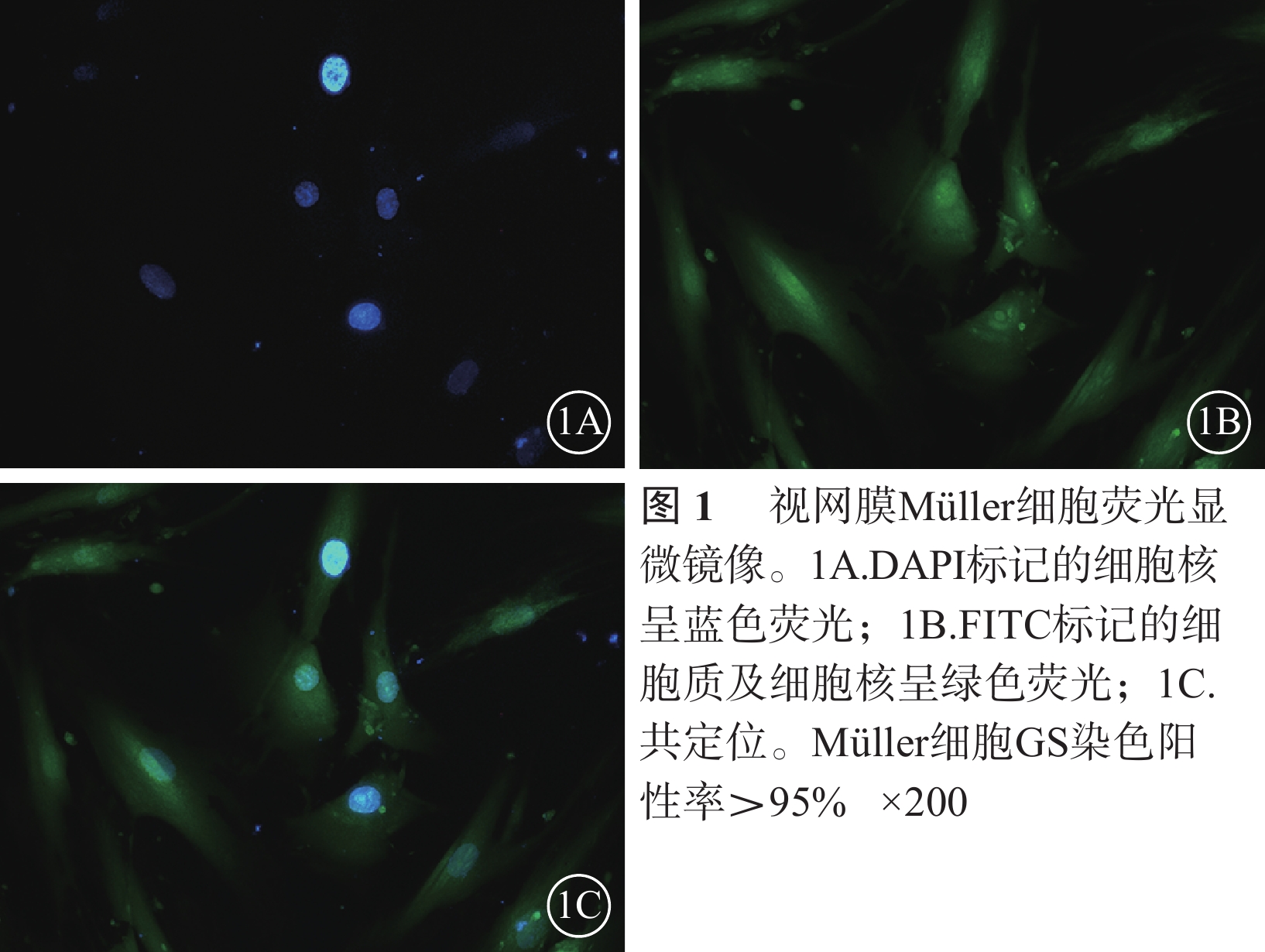
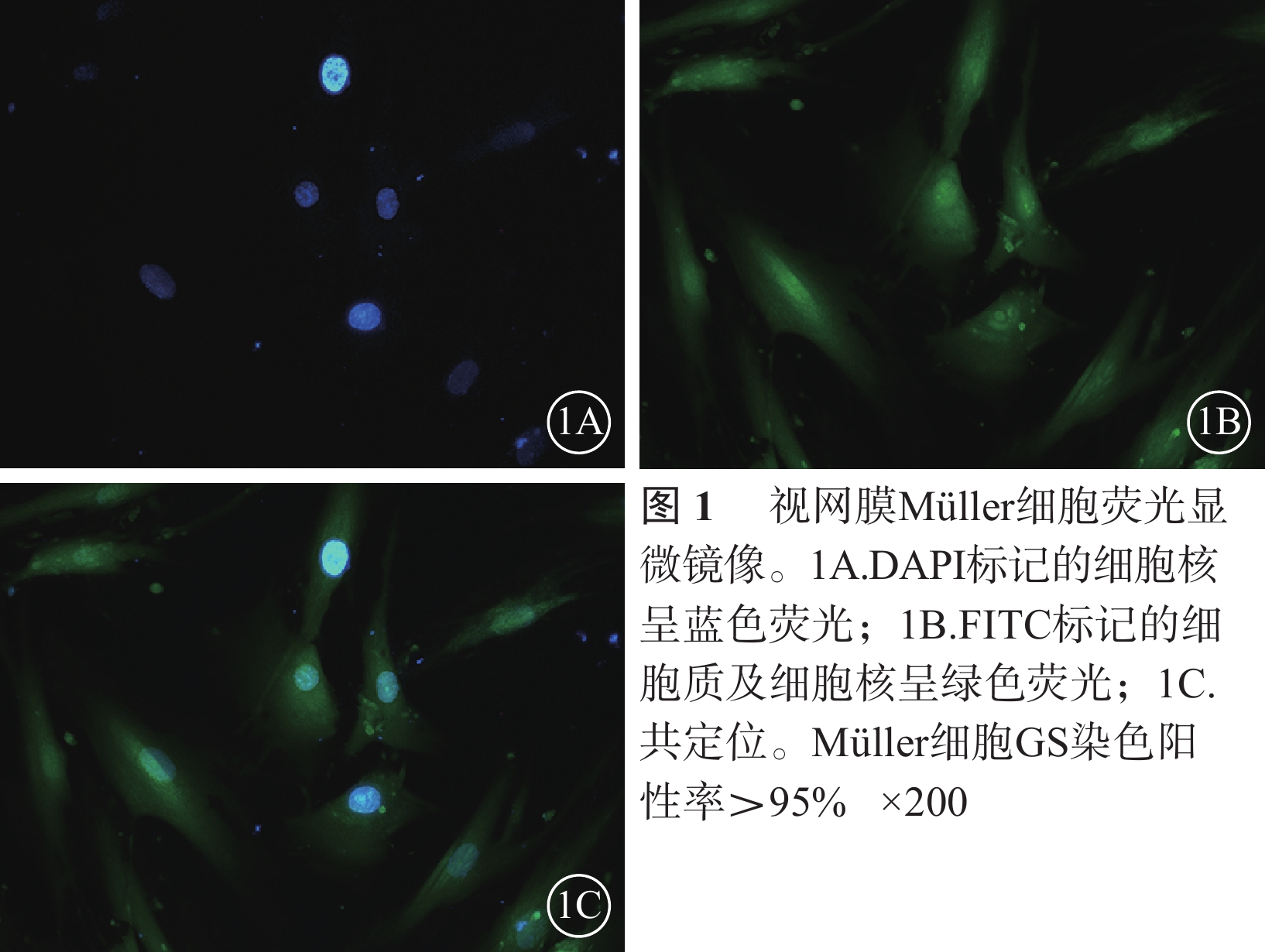
熒光顯微鏡觀察發現,95%以上Müller細胞GS染色陽性;細胞體大,細胞漿豐富,綠色熒光均勻一致;細胞核DAPI染色呈圓形或卵圓形,邊界清晰,藍色熒光均勻一致(圖1)。
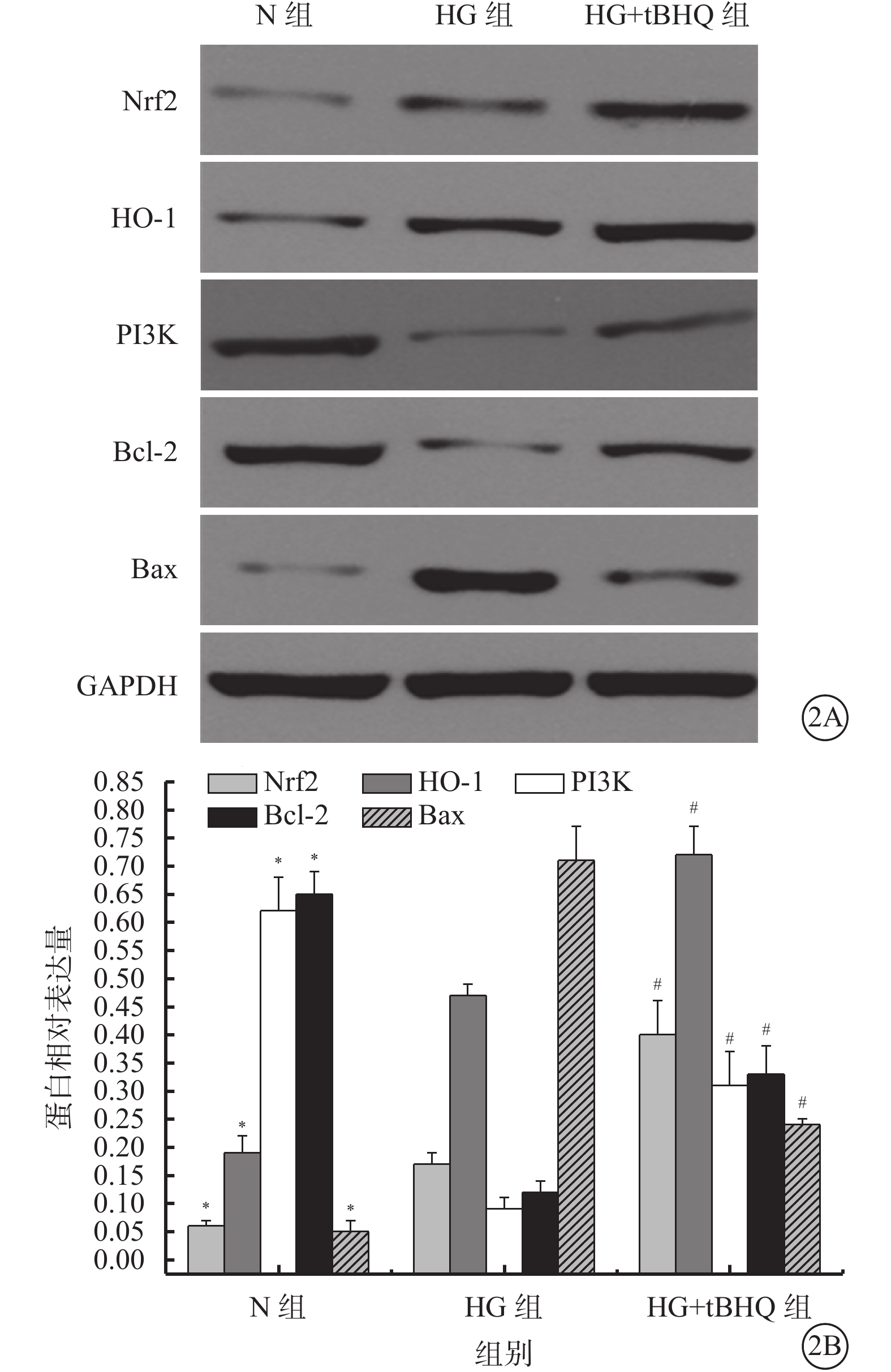
Western blot檢測結果顯示,N組、HG組、HG+tBHQ組Müller細胞中Nrf2(F=73.831)、HO-1(F=148.618)、PI3K(F=83.540)、Bcl-2(F=150.381)、Bax(F=234.459)蛋白表達比較,差異均有統計學意義(P=0.000、0.000、0.000、0.000、0.000)。 HG組Müller細胞中Nrf2(t=4.114)、HO-1(t=9.275)蛋白表達較N組升高,差異有統計學意義(P=0.006、0.000)。HG+tBHQ組Müller細胞中Nrf2(t=7.847)、HO-1(t=7.947)、PI3K(t=5.397)、Bcl-2(t=6.825)蛋白表達較HG組升高,差異有統計學意義(P=0.000、0.000、0.002、0.000);Bax蛋白表達較HG組降低,差異有統計學意義(t=14.998、P=0.000)(圖2)。
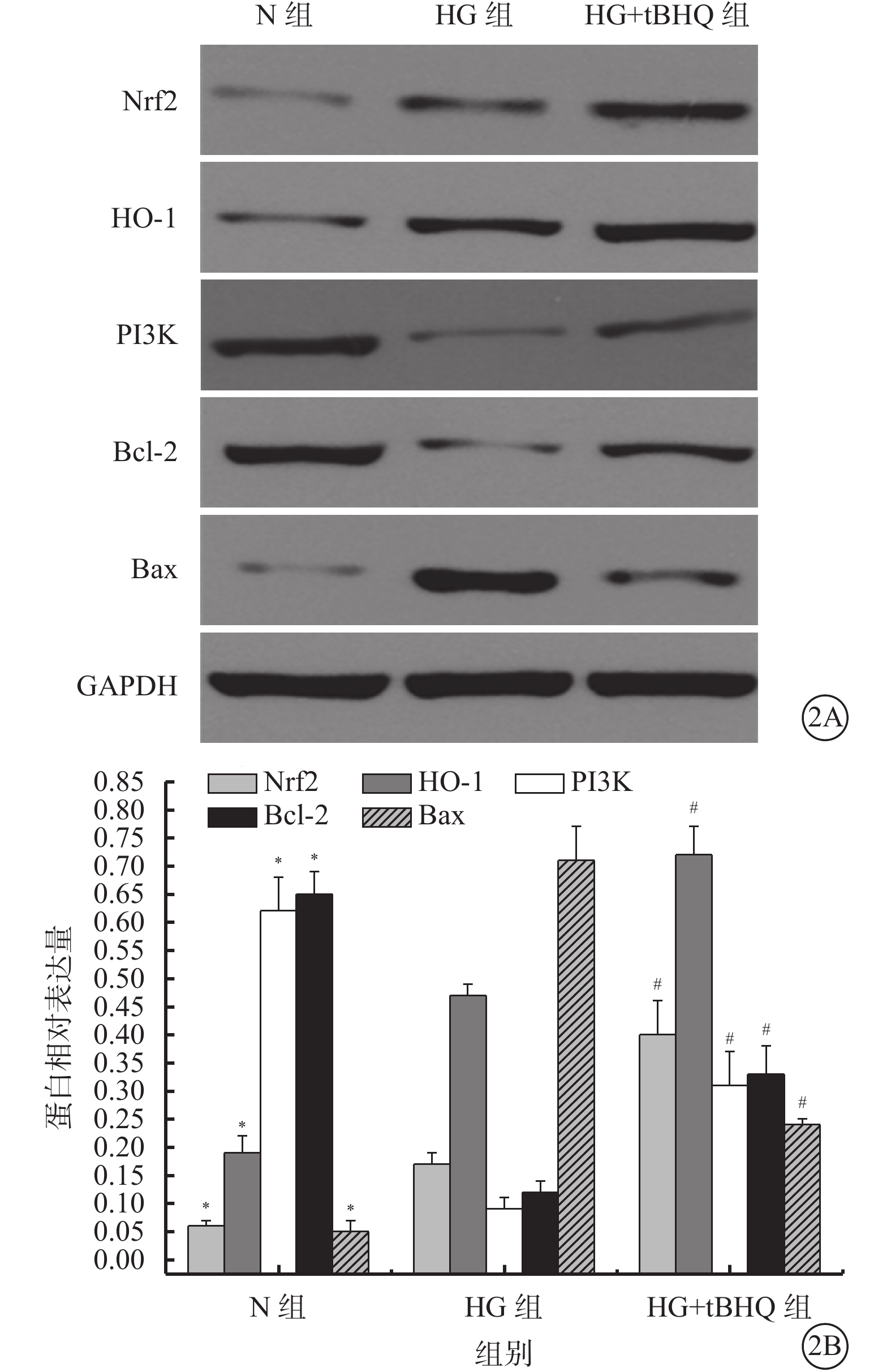 圖2
N組、HG組、HG+tBHQ組Müller細胞中Nrf2、HO-1、PI3K、Bcl-2、Bax蛋白表達情況。2A. 電泳圖;2B. Nrf2、HO-1、PI3K、Bcl-2、Bax蛋白表達結果。* HG組與N組比較,P<0.05;# HG+tBHQ組與HG組比較,P<0.05
圖2
N組、HG組、HG+tBHQ組Müller細胞中Nrf2、HO-1、PI3K、Bcl-2、Bax蛋白表達情況。2A. 電泳圖;2B. Nrf2、HO-1、PI3K、Bcl-2、Bax蛋白表達結果。* HG組與N組比較,P<0.05;# HG+tBHQ組與HG組比較,P<0.05
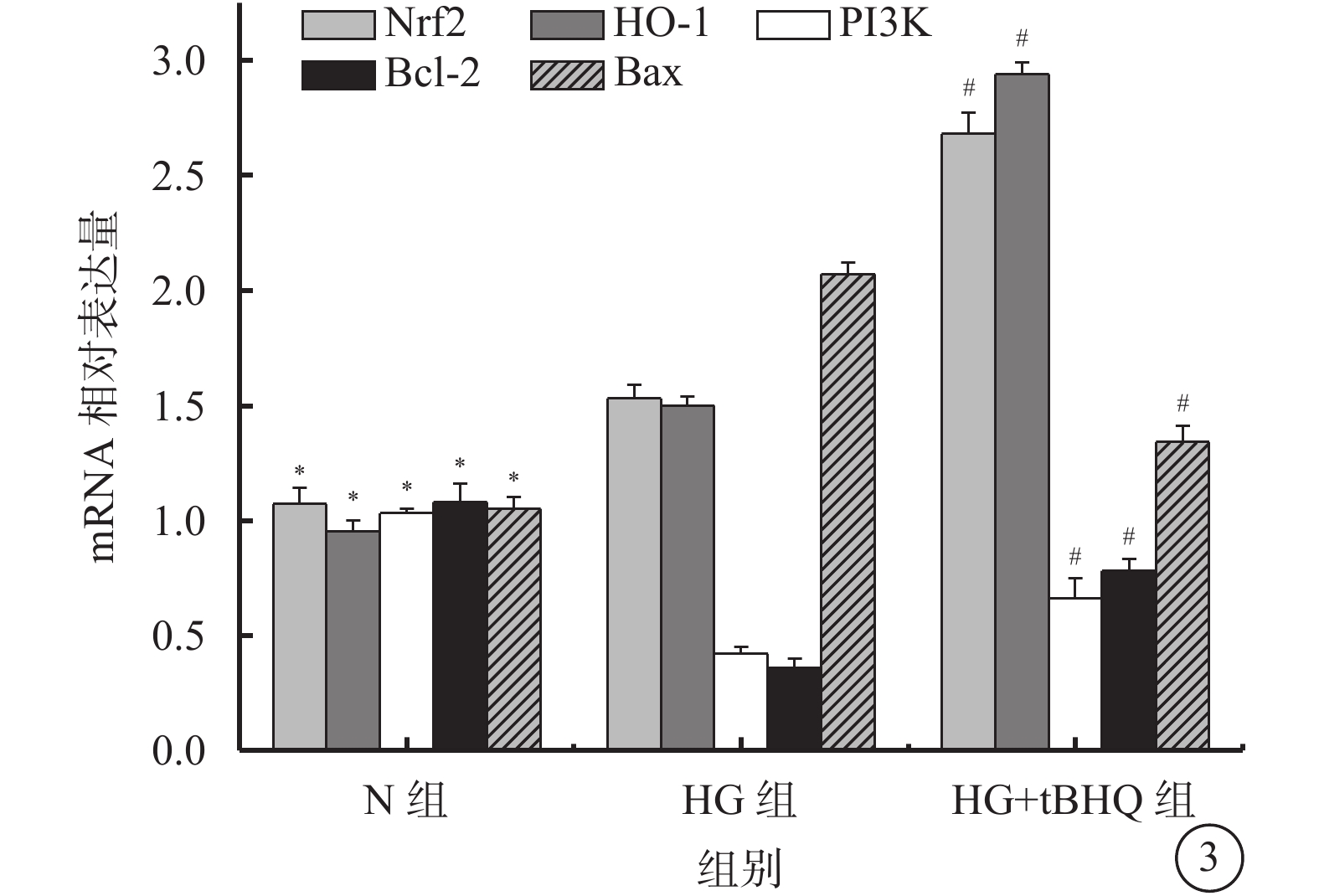
qRT-PCR檢測結果顯示,N組、HG組、HG+tBHQ組Müller細胞中Nrf2(F=340.317)、HO-1(F=1 582.911)、PI3K(F=81.807)、Bcl-2(F=133.630)、Bax(F=283.850)mRNA比較,差異有統計學意義(P=0.000、0.000、0.000、0.000、0.000)。 HG組Müller細胞中Nrf2(t=7.292)、HO-1(t=15.014)較N組升高,差異有統計學意義(P=0.000、0.000)。HG+tBHQ組Müller細胞中Nrf2(t=18.046)、HO-1(t=39.458)、PI3K(t=4.979)、Bcl-2(t=9.535)mRNA較HG組升高,差異有統計學意義(P=0.000、0.000、0.003、0.000);Bax mRNA較HG組降低,差異有統計學意義(t=16.520、P=0.000)(圖3)。
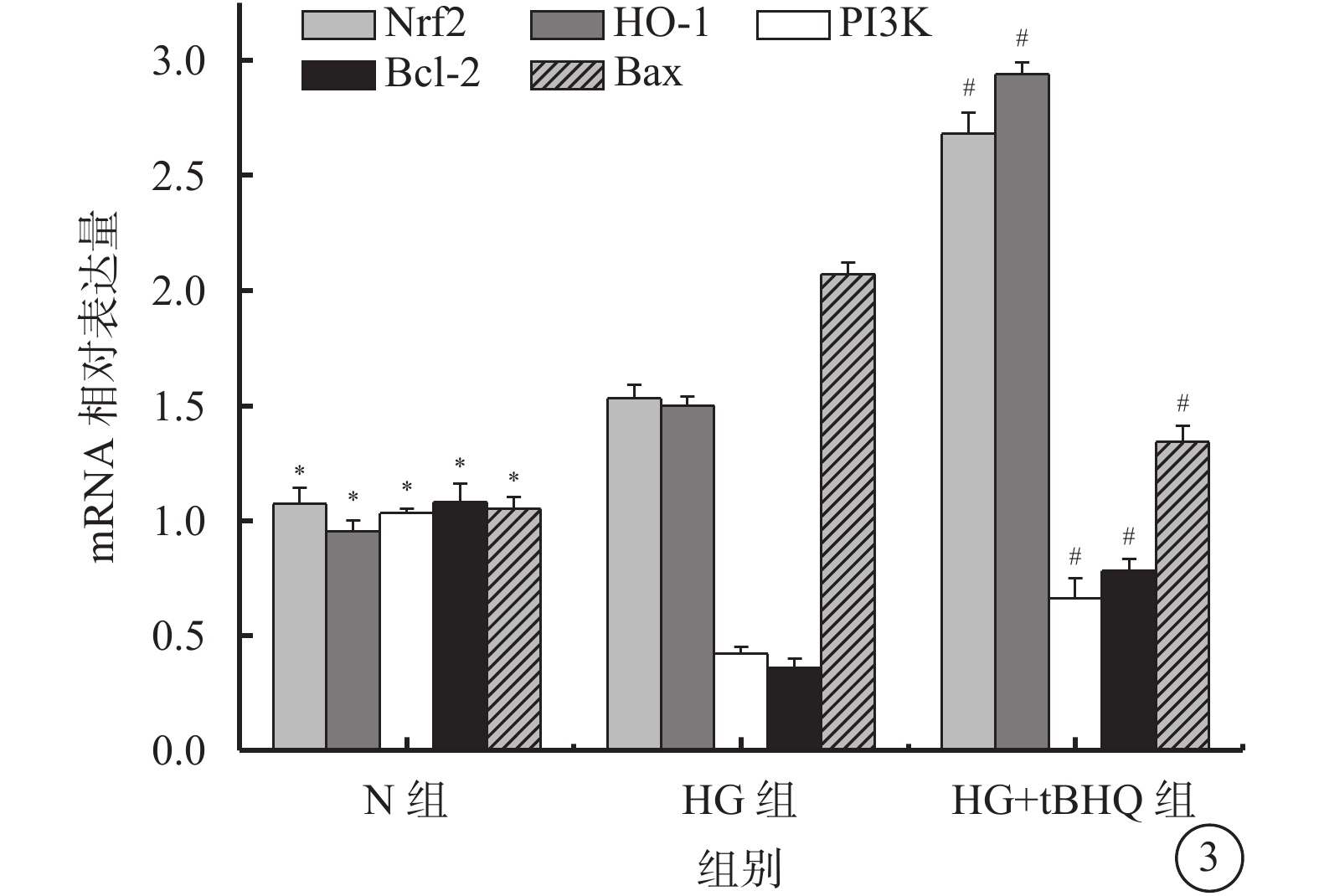 圖3
N組、HG組、HG+tBHQ組Müller細胞中Nrf2、HO-1、PI3K、Bcl-2、Bax mRNA表達情況。 * HG組與N組比較,P<0.05;# HG+tBHQ組與HG組比較,P<0.05
圖3
N組、HG組、HG+tBHQ組Müller細胞中Nrf2、HO-1、PI3K、Bcl-2、Bax mRNA表達情況。 * HG組與N組比較,P<0.05;# HG+tBHQ組與HG組比較,P<0.05
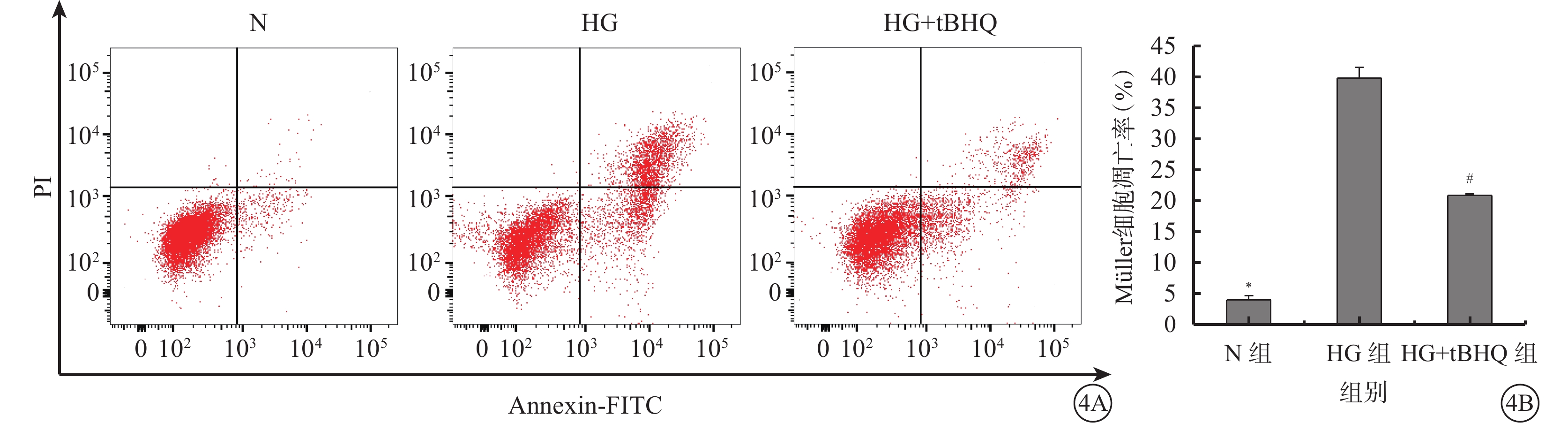
流式細胞儀檢測結果顯示,N組、HG組、HG+tBHQ組Müller細胞的凋亡率分別為(3.95±0.68)%、(39.77±1.76)%、(20.84±0.23)%。三組Müller細胞凋亡率比較,差異有統計學意義(F=797.079、P=0.000)。HG組Müller細胞凋亡率較N組增高,差異有統計學意義(t=39.905、P=0.000);HG+tBHQ組Müller細胞凋亡率較HG組降低,差異均有統計學意義(t=21.083、P=0.000)(圖4)。
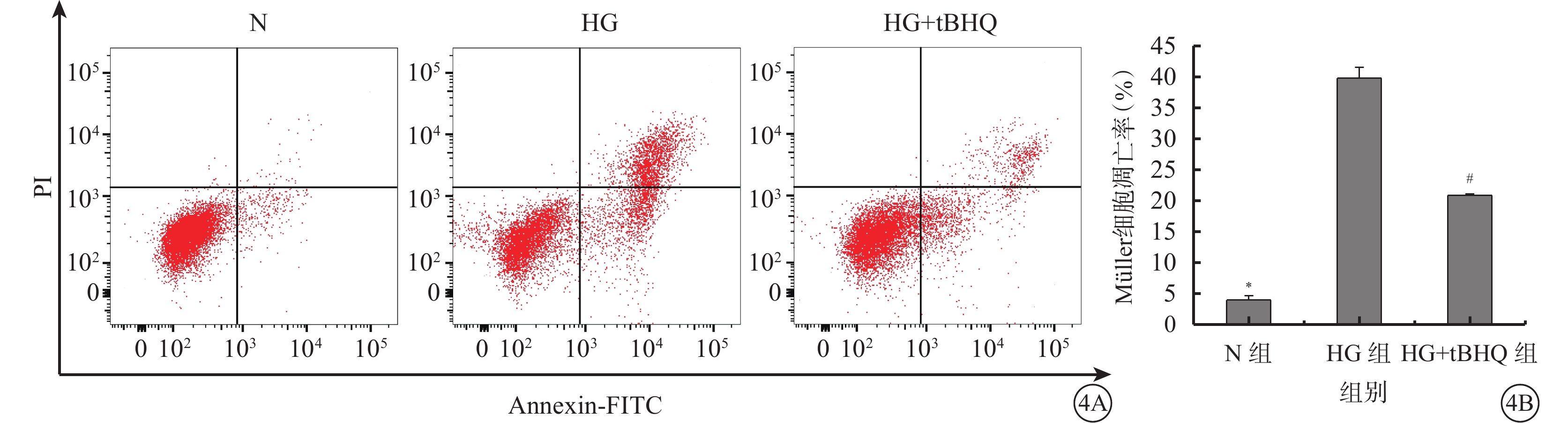 圖4
Annexin V-FITC/PI雙染法流式細胞儀檢測N組、HG組、HG+tBHQ組Müller細胞凋亡情況。4A.三組Müller細胞凋亡結果;4B.三組Müller細胞凋亡率比較。* HG組與N組比較,P<0.05;# HG+tBHQ組與HG組比較,P<0.05
圖4
Annexin V-FITC/PI雙染法流式細胞儀檢測N組、HG組、HG+tBHQ組Müller細胞凋亡情況。4A.三組Müller細胞凋亡結果;4B.三組Müller細胞凋亡率比較。* HG組與N組比較,P<0.05;# HG+tBHQ組與HG組比較,P<0.05
3 討論
目前已經發現Nrf2在人視網膜Müller細胞中表達[18],同時也發現DR患者視網膜中Nrf2表達增加[19]。本研究結果發現,N組Müller細胞中Nrf2表達,HG組Nrf2表達增加。Nrf2可調節直接和間接抗氧化,間接酶包括HO-1[20-22]。本研究結果表明,高糖條件下,Müller細胞中Nrf2及其下游抗氧化蛋白HO-1的表達增加,說明此時Müller細胞有助于維持細胞氧化還原狀態并防止氧化。既往部分研究發現,盡管糖尿病環境中Nrf2表達增加,但其在細胞核中的表達水平和DNA結合活性卻低于正常水平[19]。表明雖然在糖尿病環境中細胞產生更多的Nrf2,導致Nrf2產量增加,但是不能到達細胞核以增強轉錄機制。
本研究結果發現,tBHQ干預進一步增加了Nrf2和HO-1在Müller細胞中的表達,與Zhong等[19]結果一致。Zhong等[19]發現,tBHQ在高糖條件下干預牛視網膜內皮細胞,可以激活Nrf2活性。其他一些研究也發現,tBHQ在體內通過促進Nrf2蛋白入細胞核,繼而激活Nrf2-ARE信號通路,啟動HO-1基因的表達[23,24]。上述結果均表明,tBHQ可以通過增加Nrf2和HO-1的活性發揮Müller細胞的抗氧化功能。
PI3K/Akt通路在DR中發揮重要作用,糖尿病大鼠視網膜中PI3K,Akt-1和Akt-3活性喪失[3]。本研究結果也發現高糖環境下,大鼠Müller細胞中PI3K表達降低。PI3K/Akt途徑是細胞存活途徑的主要調節因子[4]。該途徑的激活可以導致Bcl-2家族的抗凋亡成員增加和促凋亡蛋白的水平降低[5-7]。作為Bcl-2家族成員,抗凋亡因子Bcl-2和促凋亡因子Bax在線粒體途徑中天生平衡,并且這種平衡可以決定糖尿病刺激后Müller細胞的存活或死亡[25-29]。越來越多的證據表明,高血糖導致Müller細胞死亡是通過細胞凋亡實現[30,31]。本研究結果顯示,高糖條件下Müller細胞Bcl-2表達下降,Bax表達增加;表明高糖環境下Müller細胞凋亡程序激活。 PI3K/Akt途徑也調控Nrf2介導的抗氧化功能[32,33]。已經有研究發現,PI3K參與Nrf2的解離和核轉運以影響其下游的氧化應激產物[34]。tBHQ可以刺激PI3k依賴性Akt磷酸化,上調Nrf2介導的抗氧化應答元件的活性[8,9]。本研究結果顯示,tBHQ誘導干預后,PI3K,Nrf2和Bcl- 2表達升高,Bax表達降低;提示tBHQ可能通過激活PI3K-Nrf2途徑抑制Müller細胞的凋亡。既往有研究發現,褪黑激素是一種理想的抗氧化藥物,褪黑激素在DR中通過激活Müller細胞中的PI3K/Akt-Nrf2信號通路,通過誘導HO-1的表達增強細胞抗氧化防御能力[35]。同時,我們的研究也發現,tBHQ干預后,HO-1隨著Nrf2表達的增加而增加;表明tBHQ可能通過激活PI3K-Nrf2途徑誘導HO-1的分泌,從而導致抗氧化應激和抗凋亡。同時有研究發現,tBHQ也可激活Nrf2-AREr3通路,誘導Bcl-2基因轉錄,下調Bax蛋白表達,起到抗氧化和抗凋亡的作用[36]。然而,也有部分研究發現,tBHQ確實能有效阻止肝細胞的死亡并抑制心肌細胞凋亡,但tBHQ的保護作用與Nrf2的激活無關,而與Akt的急性激活有關[37,38]。說明tBHQ的抗凋亡保護作用有可能是通過激活PI3K/Akt信號通路而起作用,而與Nrf2的激活無關。以上這些發現還需要進一步實驗進行驗證。
本研究參照文獻[15]的方法使用濃度為20 μmol/L tBHQ對高糖培養的Müller細胞干預48 h,發現tBHQ可誘導Müller細胞中各因子的變化。然而不同濃度的tBHQ及干預不同時間點可能會誘發各因子不一樣的表達,后期將進行進一步的研究。
本研究結果發現,高糖環境下Müller細胞產生更多的Nrf2和HO-1,雖然其產量增加,但不能到達細胞核以增強轉錄機制。tBHQ不僅能激活Nrf2和HO-1在Müller細胞中的表達以抗氧化反應,而且還通過激活PI3K信號通路來抑制Müller細胞的凋亡。 tBHQ也可能是通過激活PI3K-Nrf2途徑誘導HO-1分泌,從而抗氧化應激,抗凋亡作用。tBHQ在高糖環境下誘導Müller細胞中Nrf2、HO-1及PI3K的表達具有巨大的潛力,以保護Müller細胞免受氧化應激和凋亡的損傷,并且可能對未來治療DR具有重要的作用。
糖尿病視網膜病變(DR)發病機制包括氧化應激、細胞凋亡等多方面[1]。核因子E2相關因子2(Nrf2)信號通路是迄今為止發現的最為重要的內源性抗氧化應激通路,其可轉入細胞核內與抗氧化反應元件(ARE)結合并啟動血紅素氧合酶1(HO-1)等下游抗氧化因子轉錄,減輕氧化應激損傷[2]。磷脂酰肌醇-3激酶(PI3K)/蛋白激酶B(Akt)通路在DR中發揮重要作用[3],是細胞存活途徑的主要調節因子[4],其激活可以導致抗凋亡成員B淋巴細胞瘤-2(Bcl-2)增加和促凋亡蛋白Bax的水平降低[5-7]。叔丁基對苯二酚(tBHQ)為Ⅱ相酶誘導劑,是Nrf2/ARE信號通路的強誘導劑。并且也有研究發現tBHQ可以刺激PI3K依賴性Akt磷酸化,上調Nrf2介導的抗氧化應答元件的活性[8, 9]。我們前期研究發現,tBHQ能通過激活Nrf2/ARE信號通路,減輕2型糖尿病大鼠模型視網膜氧化應激損傷,減少視網膜血管增生,抑制視網膜細胞凋亡[10-13]。為此,本研究應用tBHQ預處理高糖環境下大鼠視網膜Müller細胞,觀察tBHQ對高糖環境下大鼠視網膜Müller細胞中Nrf2、HO-1、PI3K、Bcl-2、Bax表達的影響,初步探討Ⅱ相酶誘導劑tBHQ對大鼠視網膜Müller細胞的保護作用及相關機制。現將結果報道如下。
1 材料和方法
Sprague-Dawley大鼠視網膜Müller細胞由武漢生命科技股份有限公司提供。兔抗大鼠谷氨酰胺合成酶(GS)單克隆抗體、兔抗Nrf2多克隆抗體、兔抗Bcl-2多克隆抗體、兔抗甘油醛-3-磷酸脫氫酶(GAPDH)多克隆抗體(英國Abcam公司);異硫氰酸熒光素(FITC)偶聯的山羊抗兔二抗、RIPA總蛋白裂解液、BCA蛋白質濃度測定試劑盒、4’,6-二脒基-2-苯基吲哚(DAPI)(武漢阿斯本生物技術有限公司);兔抗HO-1多克隆抗體(美國Affbiotech公司);兔抗PI3K多克隆抗體(美國CST公司),兔抗Bax多克隆抗體(武漢三鷹生物技術有限公司);Trizol試劑(美國Invitrogen公司),PrimeScriptTMRT試劑盒(日本TaKaRa公司);錨定蛋白(Annexin V)-FITC細胞凋亡檢測試劑盒(天津三箭生物技術有限公司)。
參照文獻[14]的方法培養并鑒定Müller細胞,取第2~4代細胞進行實驗。按照培養液的葡萄糖濃度分為正常糖組(N組,5.5 mmol/L葡萄糖),高糖組(HG組,45.0 mmol/L葡萄糖)及tBHQ干預組(HG+tBHQ組)。參照文獻[15]方法在HG+tBHQ組視網膜Müller細胞培養48 h后,加入tBHQ 20 μmol/L干預處理48 h。
免疫熒光染色法鑒定Müller細胞,GS抗體和DAPI染色觀察Müller細胞形態。采用胰蛋白酶消化第2代對數生長期的Müller細胞,將Müller細胞用4%多聚甲醛固定在蓋玻片上15 min,磷酸鹽緩沖液(PBS)洗滌5 min。室溫下封閉工作流體處理蓋玻片10 min。PBS洗滌5 min,山羊血清室溫下封閉15 min。細胞與GS抗體在4℃溫育過夜。PBS洗滌蓋玻片,與FITC偶聯的山羊抗兔二抗在室溫下孵育50 min。PBS洗滌后,DAPI染色5 min,PBS洗滌3次,滴加適量的抗熒光淬滅劑于Müller細胞上,熒光顯微鏡下觀察。
蛋白免疫印跡法(Western blot)檢測各組Müller細胞中Nrf2、HO-1、PI3K、Bcl-2、Bax蛋白的表達。胰蛋白酶消化第2代對數生長期的Müller細胞,低速離心收集細胞,裂解液沖洗抽提蛋白,4 ℃ 13 000 g離心5 min,收集上清,每孔40 μg蛋白加上樣緩沖液后煮沸5 min,12%十二烷基硫酸鈉聚丙烯酞胺凝膠電泳,轉至聚偏氟乙烯膜,封閉。加入兔抗鼠Nrf2一抗(1∶1000),HO-1一抗(1∶2000),PI3K一抗(1∶3000),Bcl-2一抗(1∶1000)和Bax一抗(1∶1000)4 ℃孵育過夜,洗滌后加羊抗兔二抗(1∶3000),室溫孵育30 min,洗滌后加入新鮮配制的增強化學發光混合溶液到膜的蛋白面側,暗室中曝光。膠片進行掃描存檔,AlphaEaseFC軟件處理系統分析目標條帶的吸光度[A,舊稱光密度(OD)]值。以Nrf2、HO-1、PI3K、Bcl-2、Bax和GAPDH的比值作為各蛋白的相對表達量。
參照文獻[16]方法,實時熒光定量聚合酶鏈(qRT-PCR)檢測各組Müller細胞中Nrf2、HO-1、PI3K、Bcl-2、Bax mRNA的表達。Nrf2、HO-1、PI3K、Bcl-2、Bax、GAPDH引物長度分別為294、290、262、214、148、210堿基對(bp)。72~95 ℃,每上升0.5 ℃取1次熒光值,最后生成溶解曲線,所有的樣本均重復檢測3次,最后計算出Ct值。各因子的表達量采用△△Ct方法進行計算分析,即通過計算目的基因相對于參照因子的倍數來比較mRNA的表達差異,管家基因GAPDH作為內參基因。△△Ct=(待測樣品目的基因的Ct平均值?待測樣本內參基因的Ct平均值)?(對照樣品目的基因的Ct平均值)。目的基因的相對拷貝量F=2?△△Ct。
參照文獻[17]方法,流式細胞儀檢測各組Müller細胞的凋亡率。采用15 mW氬離子激光,激發光波長為488 nm,發射波長530 nm,CEL LQuest軟件自動獲取1×104個細胞樣品。流式細胞儀定量分析,計算Annexin V-FITC及碘化丙啶(PI)雙染陽性細胞百分比含量。
SPSS17.0軟件行統計學處理。結果以均數±標準差(
±s)表示。相同指標多組間比較行單因素方差分析,組間兩兩比較采用最小顯著差法檢驗。P<0.05為差異有統計學意義。
2 結果
熒光顯微鏡觀察發現,95%以上Müller細胞GS染色陽性;細胞體大,細胞漿豐富,綠色熒光均勻一致;細胞核DAPI染色呈圓形或卵圓形,邊界清晰,藍色熒光均勻一致(圖1)。
Western blot檢測結果顯示,N組、HG組、HG+tBHQ組Müller細胞中Nrf2(F=73.831)、HO-1(F=148.618)、PI3K(F=83.540)、Bcl-2(F=150.381)、Bax(F=234.459)蛋白表達比較,差異均有統計學意義(P=0.000、0.000、0.000、0.000、0.000)。 HG組Müller細胞中Nrf2(t=4.114)、HO-1(t=9.275)蛋白表達較N組升高,差異有統計學意義(P=0.006、0.000)。HG+tBHQ組Müller細胞中Nrf2(t=7.847)、HO-1(t=7.947)、PI3K(t=5.397)、Bcl-2(t=6.825)蛋白表達較HG組升高,差異有統計學意義(P=0.000、0.000、0.002、0.000);Bax蛋白表達較HG組降低,差異有統計學意義(t=14.998、P=0.000)(圖2)。
圖2
N組、HG組、HG+tBHQ組Müller細胞中Nrf2、HO-1、PI3K、Bcl-2、Bax蛋白表達情況。2A. 電泳圖;2B. Nrf2、HO-1、PI3K、Bcl-2、Bax蛋白表達結果。* HG組與N組比較,P<0.05;# HG+tBHQ組與HG組比較,P<0.05
qRT-PCR檢測結果顯示,N組、HG組、HG+tBHQ組Müller細胞中Nrf2(F=340.317)、HO-1(F=1 582.911)、PI3K(F=81.807)、Bcl-2(F=133.630)、Bax(F=283.850)mRNA比較,差異有統計學意義(P=0.000、0.000、0.000、0.000、0.000)。 HG組Müller細胞中Nrf2(t=7.292)、HO-1(t=15.014)較N組升高,差異有統計學意義(P=0.000、0.000)。HG+tBHQ組Müller細胞中Nrf2(t=18.046)、HO-1(t=39.458)、PI3K(t=4.979)、Bcl-2(t=9.535)mRNA較HG組升高,差異有統計學意義(P=0.000、0.000、0.003、0.000);Bax mRNA較HG組降低,差異有統計學意義(t=16.520、P=0.000)(圖3)。
圖3
N組、HG組、HG+tBHQ組Müller細胞中Nrf2、HO-1、PI3K、Bcl-2、Bax mRNA表達情況。 * HG組與N組比較,P<0.05;# HG+tBHQ組與HG組比較,P<0.05
流式細胞儀檢測結果顯示,N組、HG組、HG+tBHQ組Müller細胞的凋亡率分別為(3.95±0.68)%、(39.77±1.76)%、(20.84±0.23)%。三組Müller細胞凋亡率比較,差異有統計學意義(F=797.079、P=0.000)。HG組Müller細胞凋亡率較N組增高,差異有統計學意義(t=39.905、P=0.000);HG+tBHQ組Müller細胞凋亡率較HG組降低,差異均有統計學意義(t=21.083、P=0.000)(圖4)。
圖4
Annexin V-FITC/PI雙染法流式細胞儀檢測N組、HG組、HG+tBHQ組Müller細胞凋亡情況。4A.三組Müller細胞凋亡結果;4B.三組Müller細胞凋亡率比較。* HG組與N組比較,P<0.05;# HG+tBHQ組與HG組比較,P<0.05
3 討論
目前已經發現Nrf2在人視網膜Müller細胞中表達[18],同時也發現DR患者視網膜中Nrf2表達增加[19]。本研究結果發現,N組Müller細胞中Nrf2表達,HG組Nrf2表達增加。Nrf2可調節直接和間接抗氧化,間接酶包括HO-1[20-22]。本研究結果表明,高糖條件下,Müller細胞中Nrf2及其下游抗氧化蛋白HO-1的表達增加,說明此時Müller細胞有助于維持細胞氧化還原狀態并防止氧化。既往部分研究發現,盡管糖尿病環境中Nrf2表達增加,但其在細胞核中的表達水平和DNA結合活性卻低于正常水平[19]。表明雖然在糖尿病環境中細胞產生更多的Nrf2,導致Nrf2產量增加,但是不能到達細胞核以增強轉錄機制。
本研究結果發現,tBHQ干預進一步增加了Nrf2和HO-1在Müller細胞中的表達,與Zhong等[19]結果一致。Zhong等[19]發現,tBHQ在高糖條件下干預牛視網膜內皮細胞,可以激活Nrf2活性。其他一些研究也發現,tBHQ在體內通過促進Nrf2蛋白入細胞核,繼而激活Nrf2-ARE信號通路,啟動HO-1基因的表達[23,24]。上述結果均表明,tBHQ可以通過增加Nrf2和HO-1的活性發揮Müller細胞的抗氧化功能。
PI3K/Akt通路在DR中發揮重要作用,糖尿病大鼠視網膜中PI3K,Akt-1和Akt-3活性喪失[3]。本研究結果也發現高糖環境下,大鼠Müller細胞中PI3K表達降低。PI3K/Akt途徑是細胞存活途徑的主要調節因子[4]。該途徑的激活可以導致Bcl-2家族的抗凋亡成員增加和促凋亡蛋白的水平降低[5-7]。作為Bcl-2家族成員,抗凋亡因子Bcl-2和促凋亡因子Bax在線粒體途徑中天生平衡,并且這種平衡可以決定糖尿病刺激后Müller細胞的存活或死亡[25-29]。越來越多的證據表明,高血糖導致Müller細胞死亡是通過細胞凋亡實現[30,31]。本研究結果顯示,高糖條件下Müller細胞Bcl-2表達下降,Bax表達增加;表明高糖環境下Müller細胞凋亡程序激活。 PI3K/Akt途徑也調控Nrf2介導的抗氧化功能[32,33]。已經有研究發現,PI3K參與Nrf2的解離和核轉運以影響其下游的氧化應激產物[34]。tBHQ可以刺激PI3k依賴性Akt磷酸化,上調Nrf2介導的抗氧化應答元件的活性[8,9]。本研究結果顯示,tBHQ誘導干預后,PI3K,Nrf2和Bcl- 2表達升高,Bax表達降低;提示tBHQ可能通過激活PI3K-Nrf2途徑抑制Müller細胞的凋亡。既往有研究發現,褪黑激素是一種理想的抗氧化藥物,褪黑激素在DR中通過激活Müller細胞中的PI3K/Akt-Nrf2信號通路,通過誘導HO-1的表達增強細胞抗氧化防御能力[35]。同時,我們的研究也發現,tBHQ干預后,HO-1隨著Nrf2表達的增加而增加;表明tBHQ可能通過激活PI3K-Nrf2途徑誘導HO-1的分泌,從而導致抗氧化應激和抗凋亡。同時有研究發現,tBHQ也可激活Nrf2-AREr3通路,誘導Bcl-2基因轉錄,下調Bax蛋白表達,起到抗氧化和抗凋亡的作用[36]。然而,也有部分研究發現,tBHQ確實能有效阻止肝細胞的死亡并抑制心肌細胞凋亡,但tBHQ的保護作用與Nrf2的激活無關,而與Akt的急性激活有關[37,38]。說明tBHQ的抗凋亡保護作用有可能是通過激活PI3K/Akt信號通路而起作用,而與Nrf2的激活無關。以上這些發現還需要進一步實驗進行驗證。
本研究參照文獻[15]的方法使用濃度為20 μmol/L tBHQ對高糖培養的Müller細胞干預48 h,發現tBHQ可誘導Müller細胞中各因子的變化。然而不同濃度的tBHQ及干預不同時間點可能會誘發各因子不一樣的表達,后期將進行進一步的研究。
本研究結果發現,高糖環境下Müller細胞產生更多的Nrf2和HO-1,雖然其產量增加,但不能到達細胞核以增強轉錄機制。tBHQ不僅能激活Nrf2和HO-1在Müller細胞中的表達以抗氧化反應,而且還通過激活PI3K信號通路來抑制Müller細胞的凋亡。 tBHQ也可能是通過激活PI3K-Nrf2途徑誘導HO-1分泌,從而抗氧化應激,抗凋亡作用。tBHQ在高糖環境下誘導Müller細胞中Nrf2、HO-1及PI3K的表達具有巨大的潛力,以保護Müller細胞免受氧化應激和凋亡的損傷,并且可能對未來治療DR具有重要的作用。