引用本文: 黎曉新, 王寧利, 梁小玲, 徐格致, Xiao-yanLi, JennyJiao, JeanLou, YehiaHashad, 中國視網膜靜脈阻塞地塞米松玻璃體腔植入劑研究組. 地塞米松玻璃體腔植入劑治療中國患者視網膜靜脈阻塞繼發黃斑水腫的安全性和有效性:隨機、假注射對照、多中心研究. 中華眼底病雜志, 2018, 34(3): 212-220. doi: 10.3760/cma.j.issn.1005-1015.2018.03.003 復制
視網膜靜脈阻塞(RVO)是一種常見的危害視功能的視網膜疾病。在基于人群的北京眼病研究中,大北京地區40歲及以上成年人中RVO患病率約為0.7%,且隨年齡增長而增加[1]。視網膜分支靜脈阻塞(BRVO)和視網膜中央靜脈阻塞(CRVO)是最主要兩種類型,黃斑水腫(ME)是危害其視功能的主要原因[2]。目前治療RVO-ME的方法主要有玻璃體腔注射抗血管內皮生長因子(VEGF)藥物、糖皮質激素,以及BRVO格柵樣激光光凝治療。抗VEGF藥物治療對RVO-ME通常有效,但需頻繁注射且并非對所有患者有效[3]。糖皮質激素治療RVO符合發病機制[4],但主要副作用是糖皮質激素相關眼壓升高和白內障加重[5, 6]。
地塞米松玻璃體腔植入劑(DEX)是一種由NOVADUR@固態聚合物包裹的,內含糖皮質激素地塞米松眼內植入物,可持續緩慢釋放藥物且可生物降解[7]。該植入物通過22G一次性給藥器經睫狀體扁平部植入玻璃體腔[8]。靈長類動物實驗結果證實,玻璃體腔DEX植入后前2個月地塞米松濃度均維持較高水平,此后逐漸下降,且在前6個月中均可檢測到[9]。全球GENEVA研究對比觀察了DEX 0.7 mg、DEX 0.35 mg和假注射治療BRVO、CRVO繼發ME的療效,DEX 0.7 mg和0.35mg較假注射均可顯著改善黃斑中心凹視網膜厚度(CRT),并在單次植入后前3個月患者最佳矯正視力(BCVA)得到提高[6, 10]。DEX 0.7 mg治療組在治療后1周時較假注射組更可獲得3行以上的視力提高[11]。
基于GENEVA研究結果,美國和歐洲批準DEX 0.7 mg用于RVO-ME的治療。但GENEVA研究中DEX 0.7 mg治療組僅納入38例亞裔患者,且DEX 0.7 mg治療RVO-ME的安全性和有效性尚未在亞洲人群得到充分證實[10]。由于中國大陸40歲及以上成年人中原發性開角型青光眼和原發性閉角型青光眼患病率分別約為0.7%和1.4%[12],因此在中國人群中評估糖皮質激素相關眼壓升高發生率和轉歸以明確DEX的安全性顯得尤為重要。本研究旨在評估DEX 0.7 mg較假注射對照治療BRVO、CRVO繼發ME在中國患者中的安全性和有效性。
1 對象和方法
1.1 研究方法
隨機、雙盲、假注射對照、多中心(13個中心)為期6個月的3期臨床研究,此后接續為期2個月的開放標簽的延展研究,以評估DEX 0.7 mg治療RVO-ME的療效。DEX 0.7 mg單次注射的安全性和有效性在本研究的隨機階段以假注射為對照統計得出;DEX 0.7 mg的安全性進一步在后續開放標簽的延展研究中進行評估,此階段全部患者均接受DEX 0.7 mg植入治療。本研究于2012年9月至2014年5月依據中國藥物臨床試驗管理規范法規和準則在中國開展。本研究開始時,中國國內尚無已經批準上市的用于治療RVO-ME的抗VEGF藥物。本研究設計與GENEVA全球研究類似,評估DEX 0.7 mg對中國患者的療效;研究方案經各中心獨立的倫理委員會批準通過,全部患者均簽署書面知情同意書。本研究注冊于www.ClinicalTrials.gov,注冊號為NCT01660802。
1.2 患者選擇
患者選擇評估包括篩選訪視(第?14~?1天)和基線訪視(第1天)。年齡≥18歲;熒光素眼底血管造影(FFA)檢查評估為非缺血型BRVO或CRVO;光相干斷層掃描(OCT)檢查可見黃斑區視網膜增厚并累及中心凹;BRVO、CRVO繼發ME病程分別在篩選訪視前6~12個月和6~9個月;患者視力下降與ME相關;BCVA≥34個字母且≤68個字母(對應的Snellen視力分別為20/200和20/50)[13]。BCVA檢查采用早期治療糖尿病視網膜病變研究組視力表進行。OCT檢查采用德國Heidelberg公司Spectralis OCT儀,CRT≥320 μm,或德國Zeiss公司Cirrus OCT儀,CRT≥300 μm。篩選訪視時觀察黃斑中心凹1 mm范圍內CRT厚度。
主要排除標準:缺血型RVO,定義為FFA上無灌注區累及黃斑中心凹且超過10個視盤面積;有青光眼病史;基線前3個月內接受過玻璃體腔注射抗VEGF藥物或糖皮質激素治療;基線前3個月內接受過激光光凝或內眼手術治療;經研究者評估,患者存在影響BCVA提高15個字母以上或可能影響ME或BCVA的其他眼部情況,如嚴重黃斑缺血、黃斑前膜或黃斑中心凹萎縮;篩選訪視時患者屈光間質混濁影響臨床或眼底彩色照相評估,包括但不限于視網膜前或玻璃體積血、晶狀體混濁;致密的黃斑區積血且眼底紅光反射消失。其他重要排除標準:既往接受過經平坦部玻璃體切割手術;任一眼存在活動性細菌、病毒、寄生蟲或真菌感染;任一眼曾因接受糖皮質激素治療而發生眼壓升高;基線前1個月內曾接受糖皮質激素口服、靜脈、肌內注射、硬膜外、直腸內或皮膚外用治療;基線前3個月內曾接受免疫抑制劑、免疫調節劑、抗代謝藥和(或)氮芥類藥物治療;基線前2周曾接受眼局部糖皮質激素點眼或中藥治療;對側眼BCVA<34個字母;基線訪視BCVA較篩選訪視提高>10個字母;經研究者評估可能影響研究結果的其他因素。
1.3 治療
基線訪視時(第1天),患者按1:1隨機分為DEX治療組、假注射組,分別接受DEX 0.7 mg或假注射治療。治療組再分別按RVO類型分為BRVO或CRVO亞組。基線訪視評估后接受研究方案的相應治療。DEX治療組,DEX 0.7 mg由一次性給藥器注射植入玻璃體腔[8];假注射組,注射過程使用不帶針頭的給藥器按壓結膜。
患者在治療后第6個月時接受評估判斷是否可進入第二階段研究。若患者BCVA<84個字母(約相當于Snellen視力20/20),仍殘余ME(CRT>250 μm、視網膜內囊腔及黃斑中心區域內外視網膜厚度增加),患者可接受再次治療,且研究者評估再治療過程不會把患者置于重大風險之中。再治療時DEX治療組和假注射組均使用DEX 0.7 mg。
挽救治療。治療≥3個月后,若患者BCVA在連續兩次間隔4周的復診時,因RVO-ME加重而導致視力較基線下降≥10個字母,行挽救性激光光凝治療。任何情況下,若研究者認為挽救性激光光凝對患者有益時均可給予挽救治療。接受挽救性激光光凝治療的患者仍保留在研究中且可從第6個月開始接受開放標簽的DEX注射治療。
1.4 隨訪和評估
治療后1~8個月,每月隨訪一次。安全性評估由研究者在治療和再治療后第1天時進行。每月隨訪時均進行有效性評估,包括BCVA和OCT。篩選訪視和第6個月時對FFA檢查所見熒光素滲漏程度進行評估。讀片中心處于盲態的讀片師對FFA所示新生血管是否存在和面積大小通過對照標準圖片進行判讀。每次訪視時的主要安全性評估內容包括裂隙燈顯微鏡、Goldmann壓平眼壓計、OCT檢查等以及治療相關不良事件(TEAEs)的發生情況。研究者使用裂隙燈顯微鏡,對照年齡相關眼部疾病研究臨床晶狀體分級系統的標準圖片,對患者晶狀體核、皮質和后囊下混濁存在與否及嚴重程度進行分級[14]。TEAEs定義為基線之后發生或嚴重程度加重的不良事件,及任何嚴重不良事件。全部患者、對患者每月進行隨訪評估的研究者、收集有效數據的研究人員和讀片中心(美國Doheny圖像閱讀中心)對OCT圖像進行評估的讀片師均對患者的分組情況處于盲態。
主要有效終點為治療后6個月中患者BCVA較基線提高≥15個字母所需的時間。關鍵次要終點為治療后6個月每次隨訪時BCVA較基線提高≥15個字母的患者百分比,以及OCT檢查所示CRT較基線的變化。治療后6個月內患者BCVA較基線的變化值通過曲線下面積(AUC)的方式進行評估。依據RVO診斷(BRVO或CRVO)的亞組分析也作為重要評價指標。
1.5 數據分析和統計方法
由全部接受隨機分組和治療的患者組成的修正治療人群(mITT)用于主要有效終點的分析。全部遵守和無嚴重違反研究規程且接受隨機分組和治療的患者均用于主要終點的輔助性分析。依據患者真實接受的治療,對由全部接受治療的患者組成的安全性人群的數據進行評估得出安全性數據。
采用SAS 9.3軟件進行統計分析,雙側檢驗α=0.05。接受挽救治療之后的患者數據不用于任何有效性分析,且挽救治療之后的有效性數據均設置為缺失,用末次觀察轉結法代替。
采用Kaplan-Meier生存曲線法分析治療后前6個月患者BCVA較基線提高≥15個字母所需的時間。其中,治療后6個月無反應的患者將從第6個月時被剔除;若患者在前6個月內退出研究,則該時間點設定為末次接受視力測定的時間。BCVA提高≥15個字母之前接受挽救治療的患者也在接受初次挽救治療的時間點被剔除。治療組間的累積有效率用秩和檢驗進行比較。
治療后6個月內平均BCVA通過AUC方法計算得出。AUC通過梯形面積疊加法,以BCVA和測量時的研究天數計算得出。DEX治療組、假注射組患者之間平均BCVA比較行雙因素方差分析(ANOVA),其中治療分組和RVO類型(BRVO或CRVO)均被設定為固定因素。其他全部關于BCVA和OCT數據的次要分析,連續變量使用ANOVA法,分類變量使用Mantel-Haenszel法依據RVO類型(BRVO或CRVO)分層分析。TEAEs按照MedDRA第17.0版推薦用詞編碼,與白內障相關的全部TEAEs(白內障、糖尿病性白內障、核性白內障、囊下性白內障、皮質性白內障或晶狀體核混濁)均被評估。
預計每組樣本量為130例患者時,BCVA在治療后前6個月較基線提高≥15個字母的時間兩組存在差異的效力為85%,假定假注射組的累積反應為22.5%,DEX組相對假注射組的固定風險比為2。
2 結果
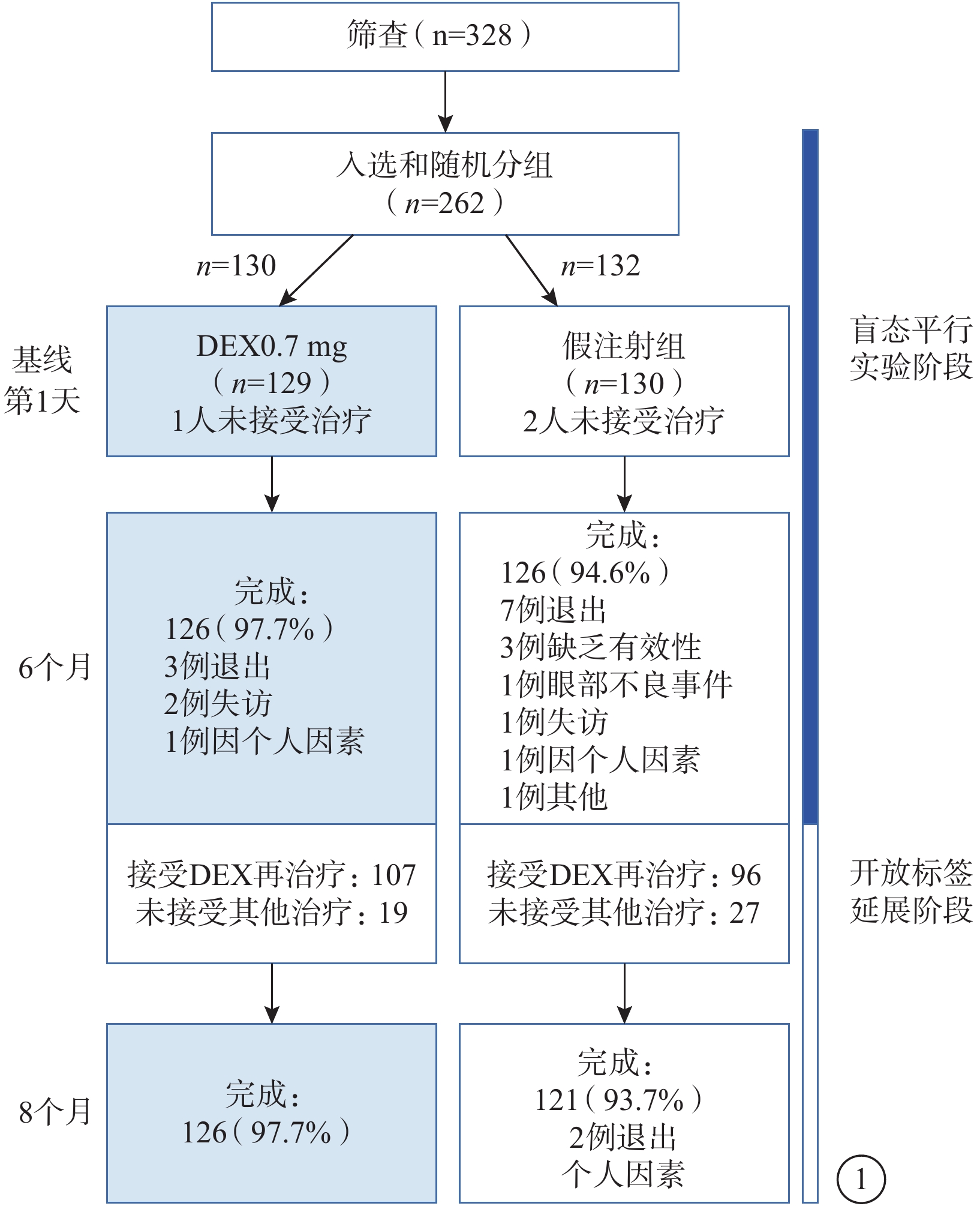
共328例患者接受篩選訪視,其中262例患者進入研究并隨機分組接受DEX或假注射治療。納入研究并隨機分組的262例患者中,因未接受治療而從mITT人群中剔除3例(DEX治療組1例、假注射組2例)。因此,共計259例患者259只眼納入最終分析(表1)。所有患者均為亞裔。DEX治療組男性患者所占比例(53.5%)較假注射組(41.5%)更大,但兩組患眼疾病特征無差異。259只眼中,255只眼(98.5%)為有晶狀體眼;BRVO、CRVO約各占一半。DEX治療組、假注射組前6個月雙盲階段研究的完成率分別為97.7%、94.6%。假注射組中1例患者因不良事件而退出研究,為黃斑囊樣水腫者(圖1)。
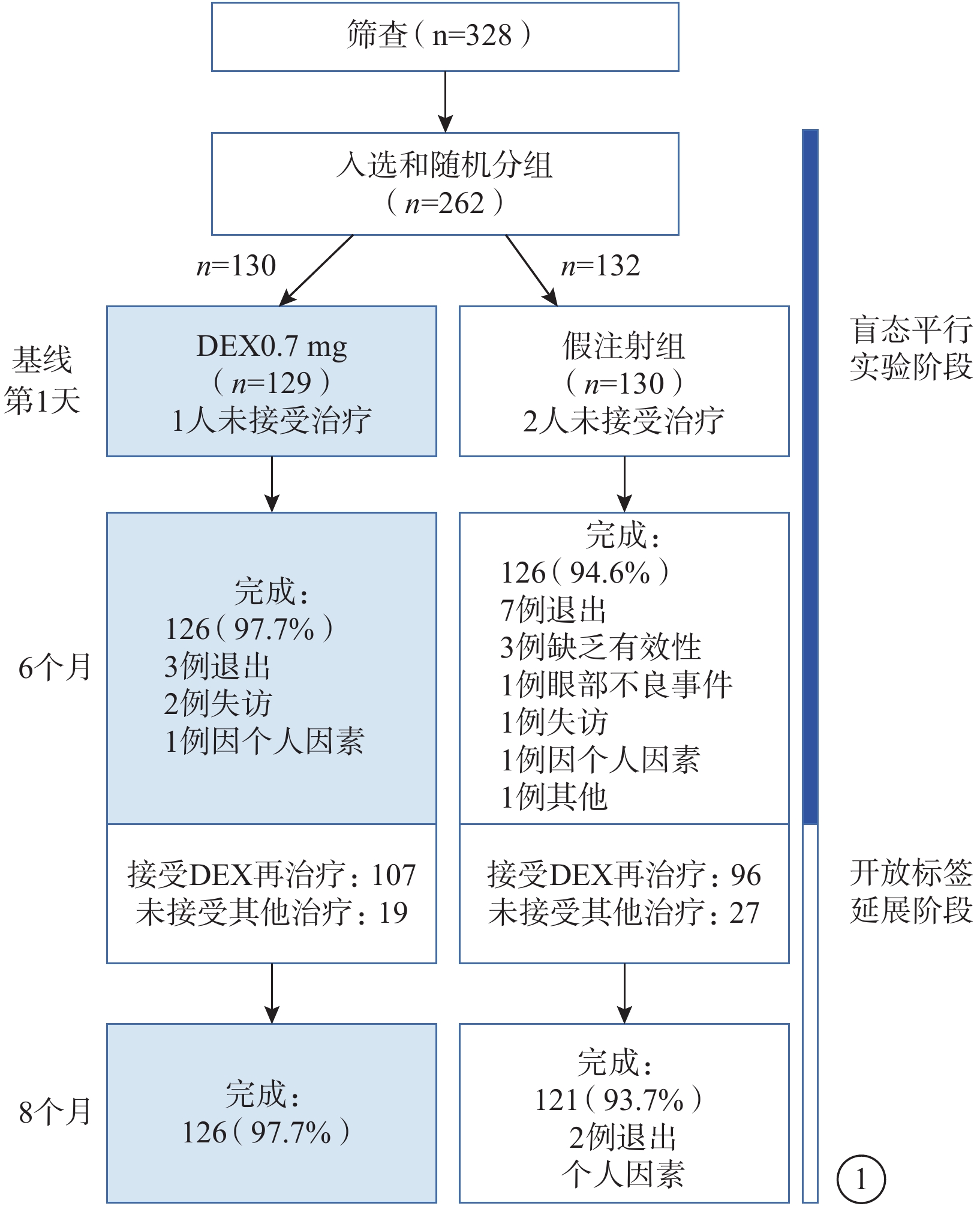 圖1
本研究患者入組流程圖(mITT人群)
圖1
本研究患者入組流程圖(mITT人群)
第6個月時,共203例患者進入后續開放標簽的DEX注射延展治療階段,包括DEX治療組107例(BRVO 53例、CRVO 54例)及假注射組96例(BRVO 41例、CRVO 55例)。249例完成前段研究的患者中,有46例患者(BRVO 30例、CRVO 16例)在第6個月時不再接受治療;其中,25例(54.3%)因CRT≤250 μm而不再需要治療。DEX治療組中100%(107/107)的患者均接受了再次DEX注射治療;而假注射組中99%(95/96)的患者在第6個月時接受DEX注射治療并完成后續為期2個月的開放標簽延展研究。
DEX治療組(BRVO、CRVO各7例)中10.9%(14/129)的患者和假注射組(BRVO 9例、CRVO 2例)中8.5%(11/130)的患者接受視網膜激光光凝挽救治療。DEX治療組中71.4%(10/14)的患者和假注射組中54.5%(6/11)的患者初次激光治療時間為第6個月。
2.1 有效性結果
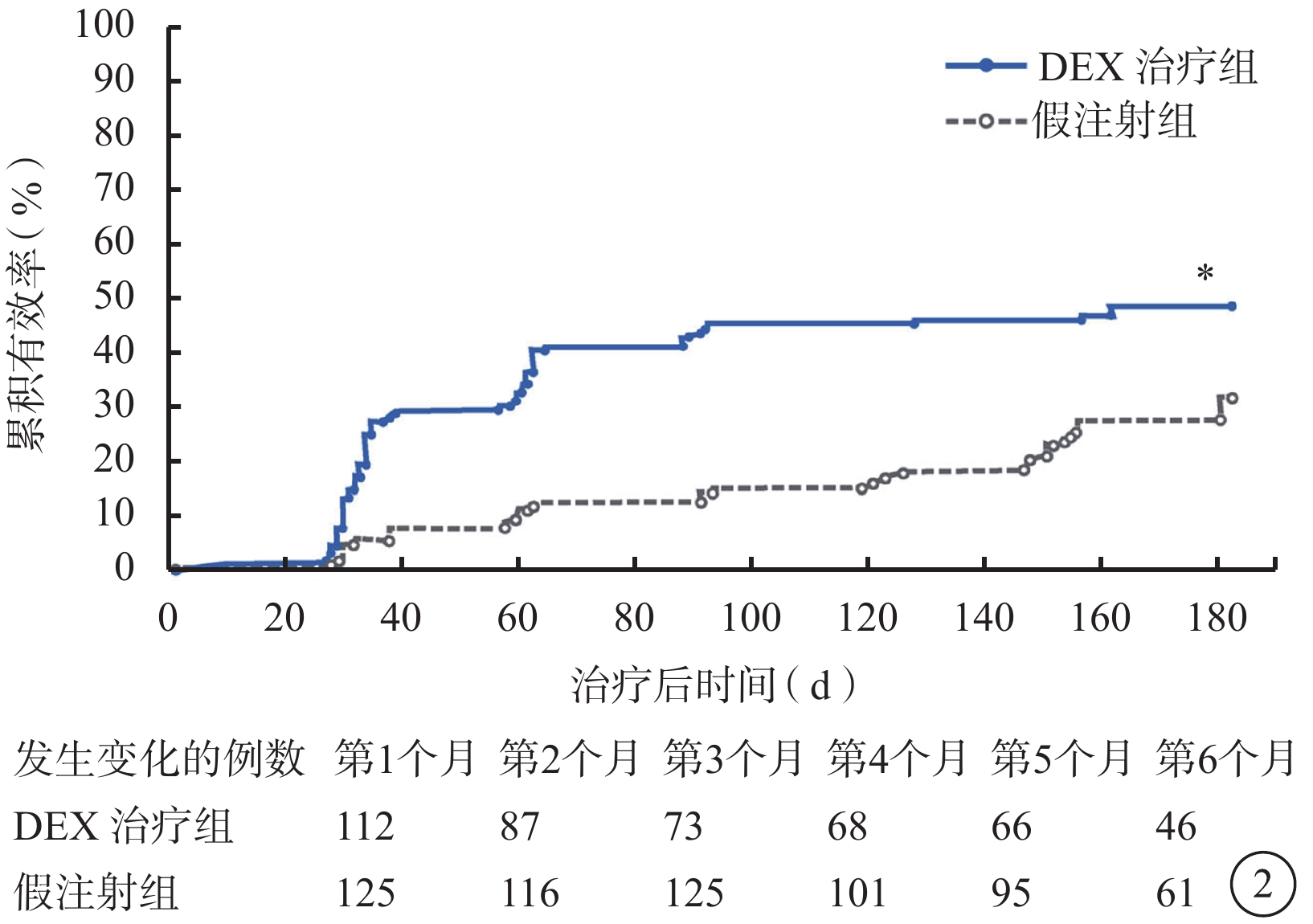
DEX可快速提高BCVA。生存曲線分析結果顯示,DEX治療組BCVA提高≥15個字母所需時間顯著短于假注射組(P<0.001)(圖2)。DEX治療組和假注射組累積有效曲線從第1次隨訪(第1個月)時即開始分離并持續至第6個月。經Cox回歸模型依據基線時RVO類型、年齡和性別調整后對全部完成隨訪的患者進行分析,證實DEX治療組和假注射組總體治療有效率存在顯著差異(P<0.001)。從該模型計算出的估計風險比為2.4(95%可信區間1.6~3.7),即DEX治療組患者獲得視力提高≥15個字母的概率是假注射組的2.4倍。
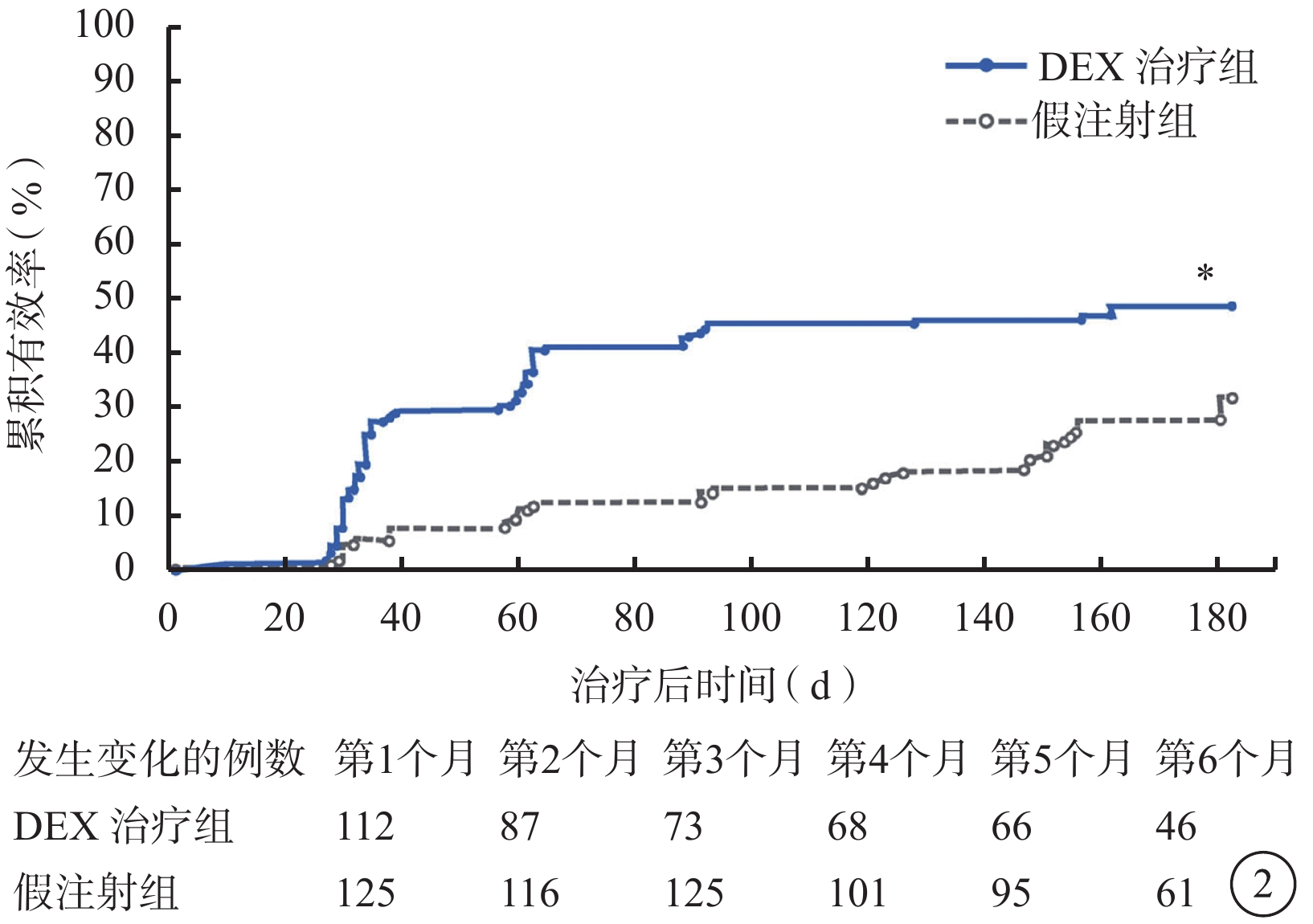 圖2
BCVA較基線提高≥15個字母所需時間的Kaplan-Meier分析圖(mITT人群)。*與假注射組比較,P<0.001
圖2
BCVA較基線提高≥15個字母所需時間的Kaplan-Meier分析圖(mITT人群)。*與假注射組比較,P<0.001
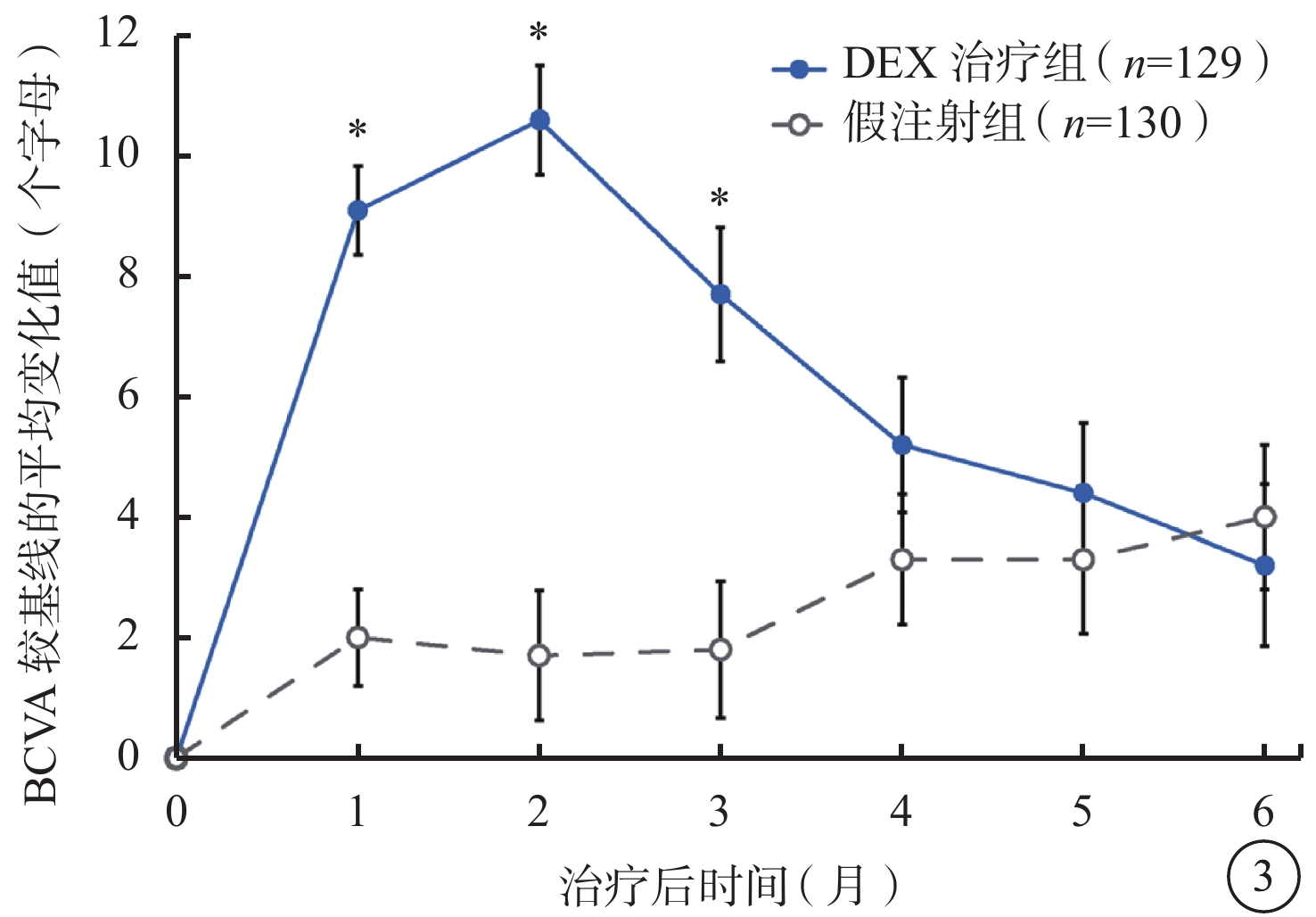
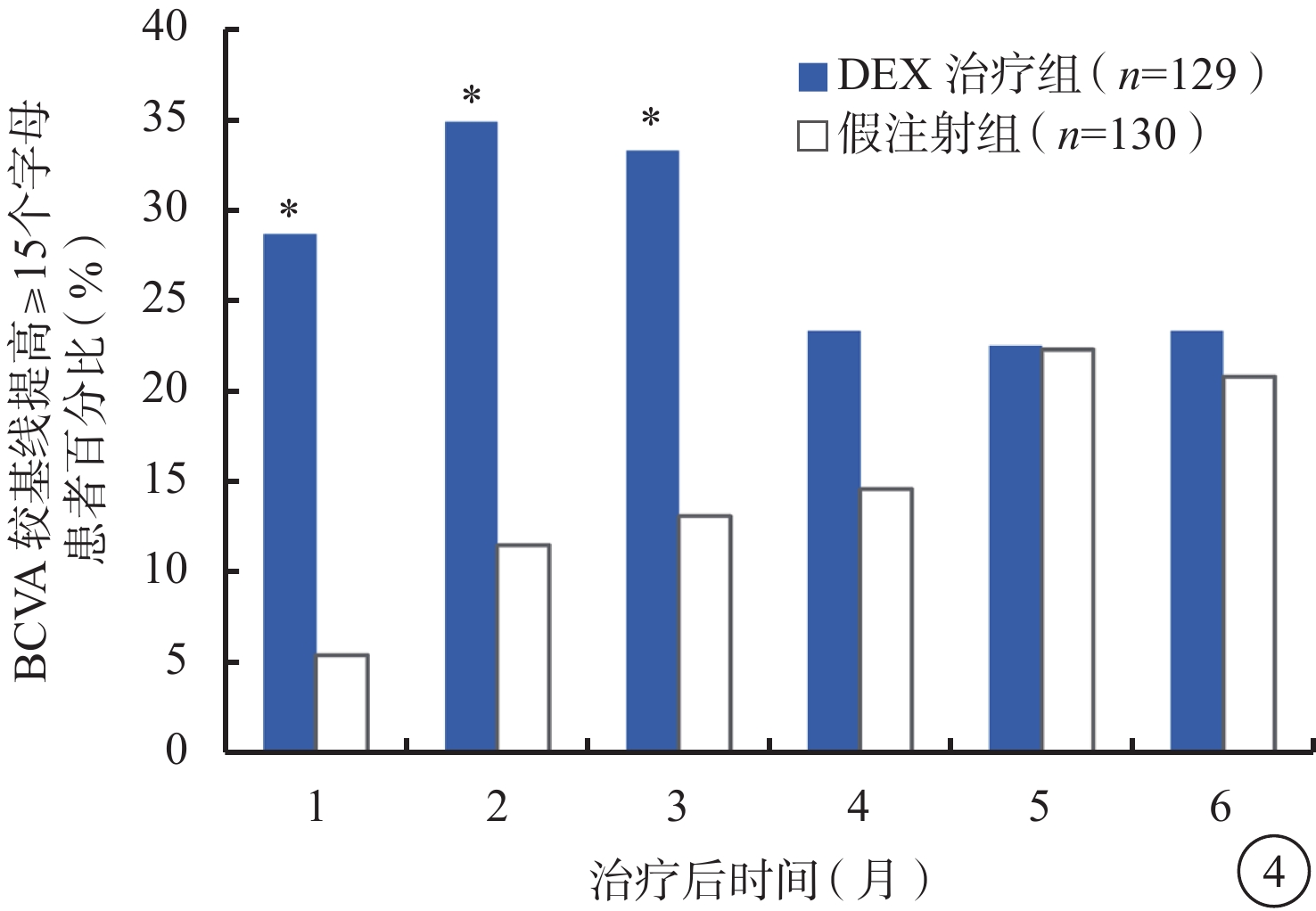
DEX治療組中BCVA較基線的變化值和BCVA較基線提高≥15個字母的患者百分比在第1、2、3個月時均顯著高于假注射組(P<0.001)(圖3,4)。第2個月時(效應峰值),DEX治療組BCVA較基線平均提高(10.6±10.4)個字母,假注射組BCVA較基線平均提高(1.7±12.3)個字母(P<0.001);DEX治療組、假注射組BCVA較基線提高≥15個字母患者百分比分別為34.9%、11.5%(P<0.001)。DEX治療組、假注射組前6個月BCVA較基線變化平均值為(6.7±9.0)、(2.5±10.0)個字母(P<0.001)。
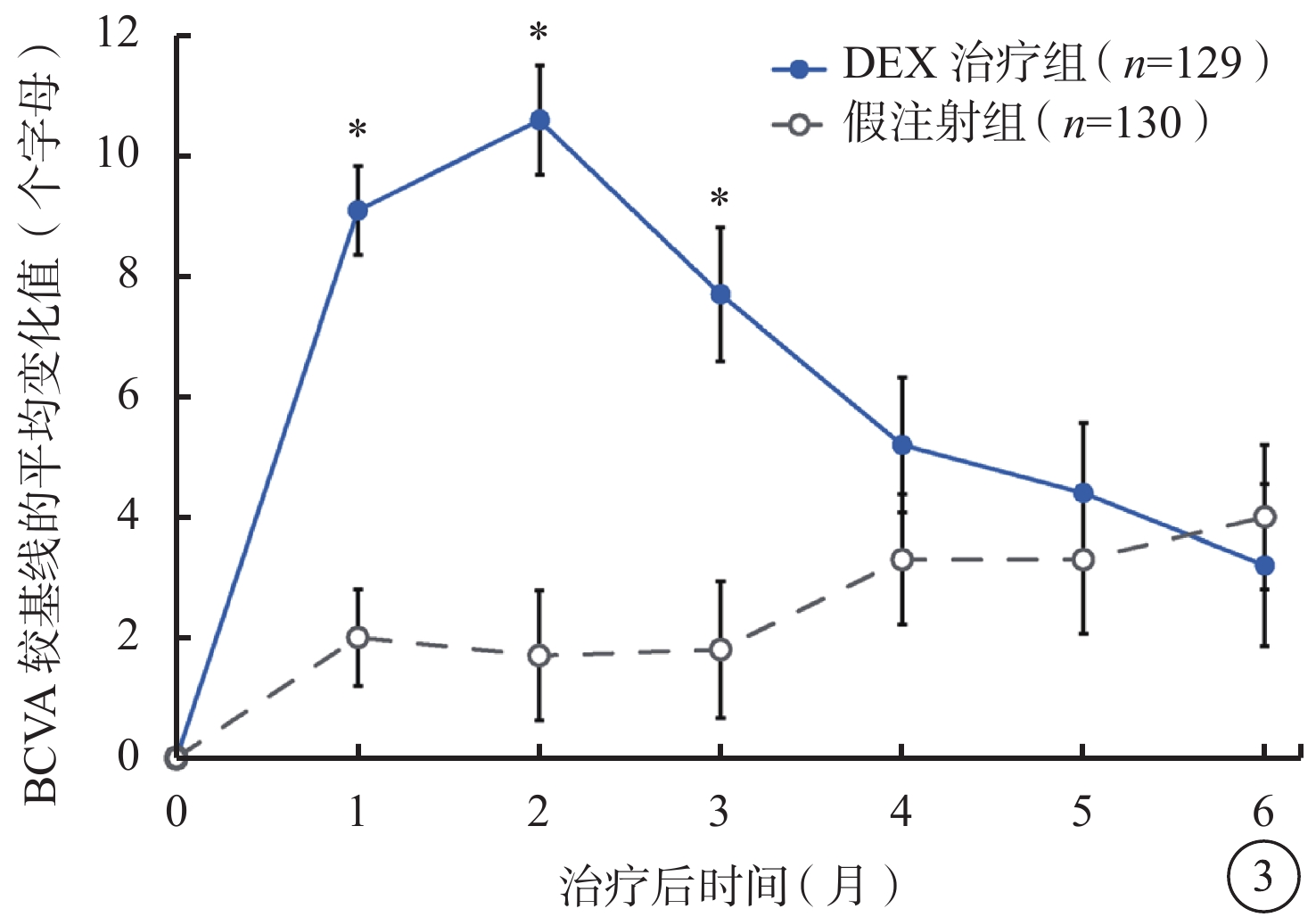 圖3
BCVA較基線的平均變化值(mITT人群)。*與假注射組比較,P<0.001
圖3
BCVA較基線的平均變化值(mITT人群)。*與假注射組比較,P<0.001
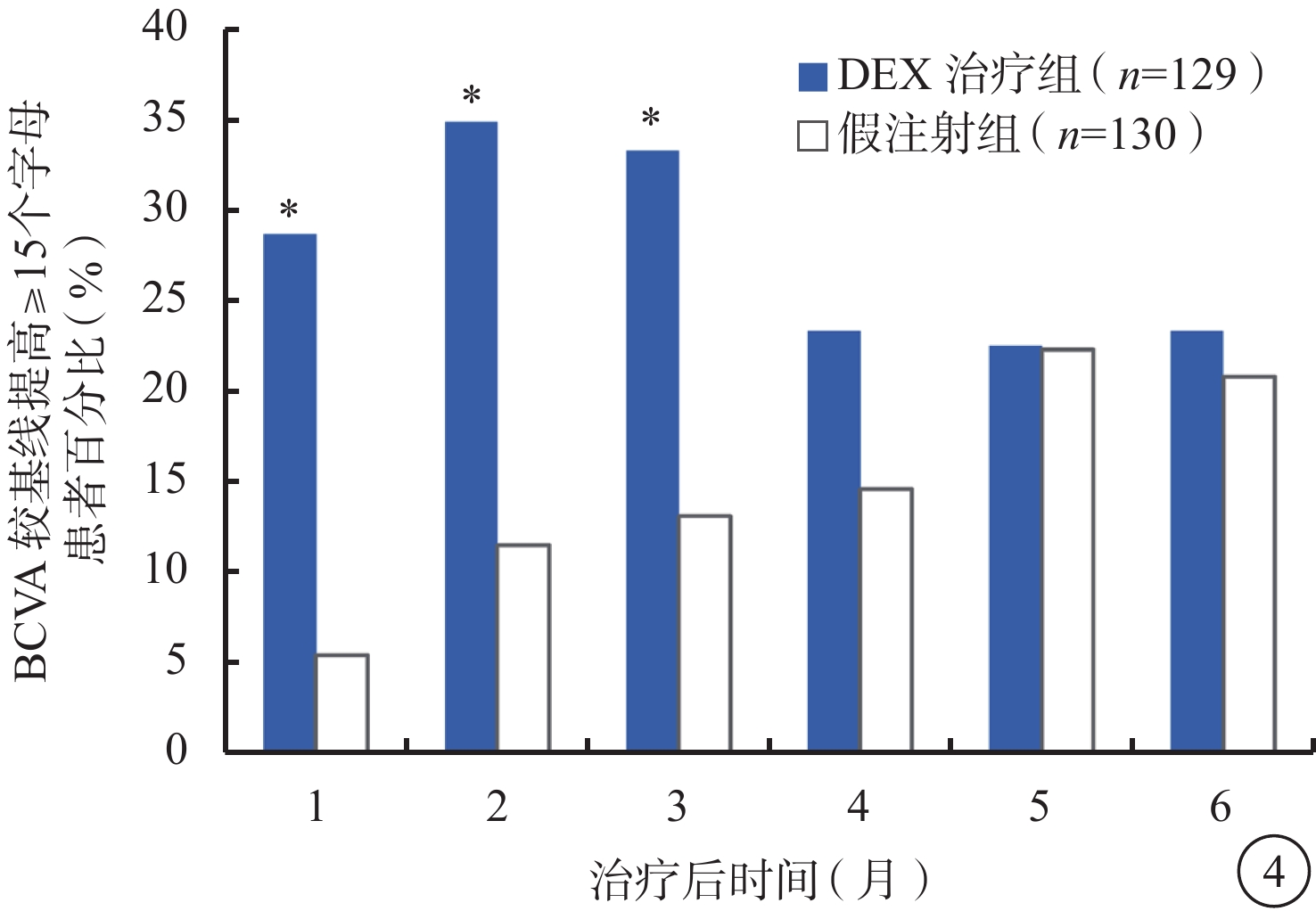 圖4
BCVA較基線提高≥15個字母患者百分比(mITT人群)。*與假注射組比較,P<0.001
圖4
BCVA較基線提高≥15個字母患者百分比(mITT人群)。*與假注射組比較,P<0.001
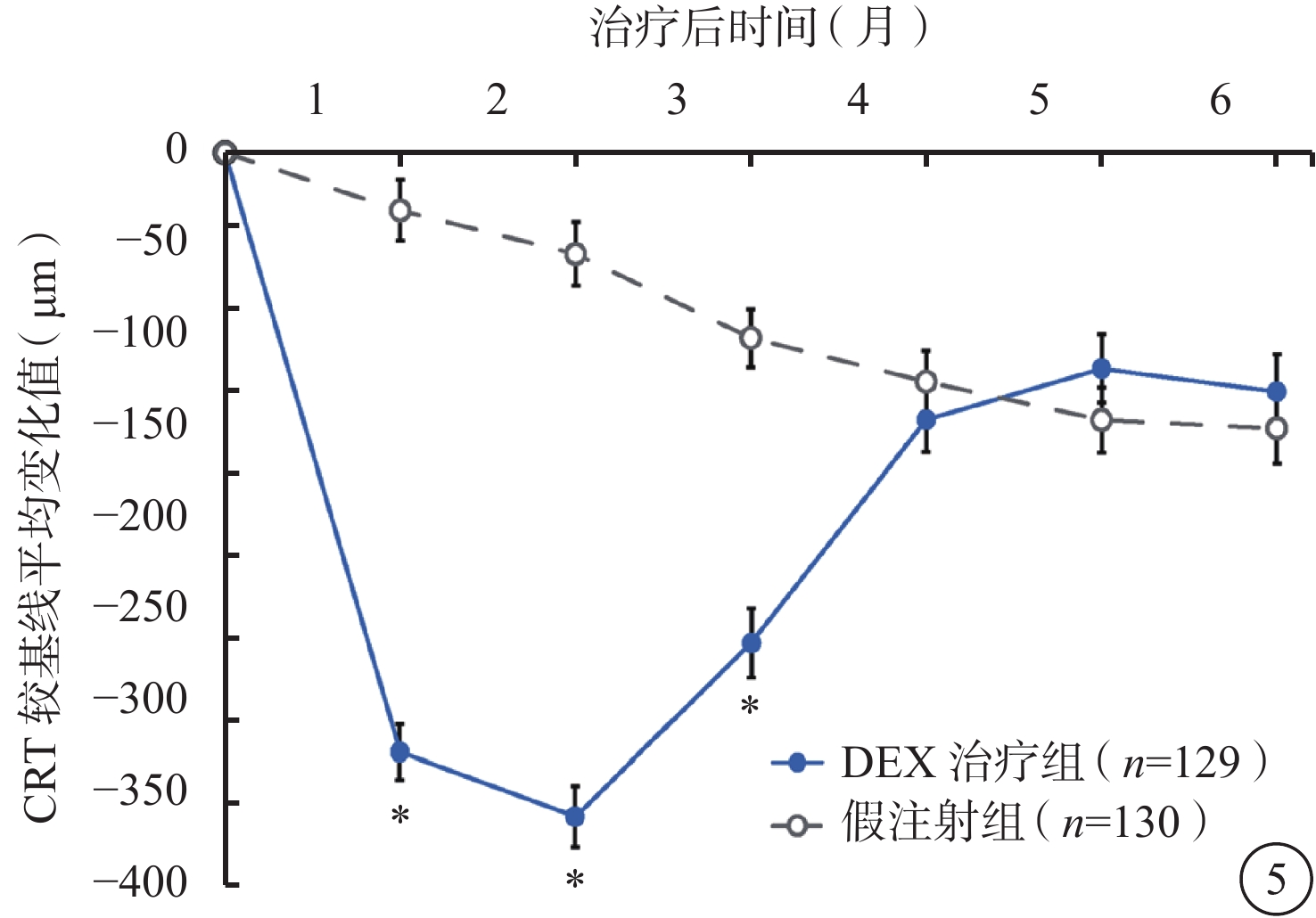
DEX治療組的解剖預后優于假注射組。第1、2、3個月時,DEX治療組CRT較基線的平均下降值顯著高于假注射組(P<0.001)(圖5)。第2個月時(效應峰值),DEX治療組CRT較基線平均降低(407±212) μm,假注射組CRT較基線平均降低(62±224)μm(P<0.001)。
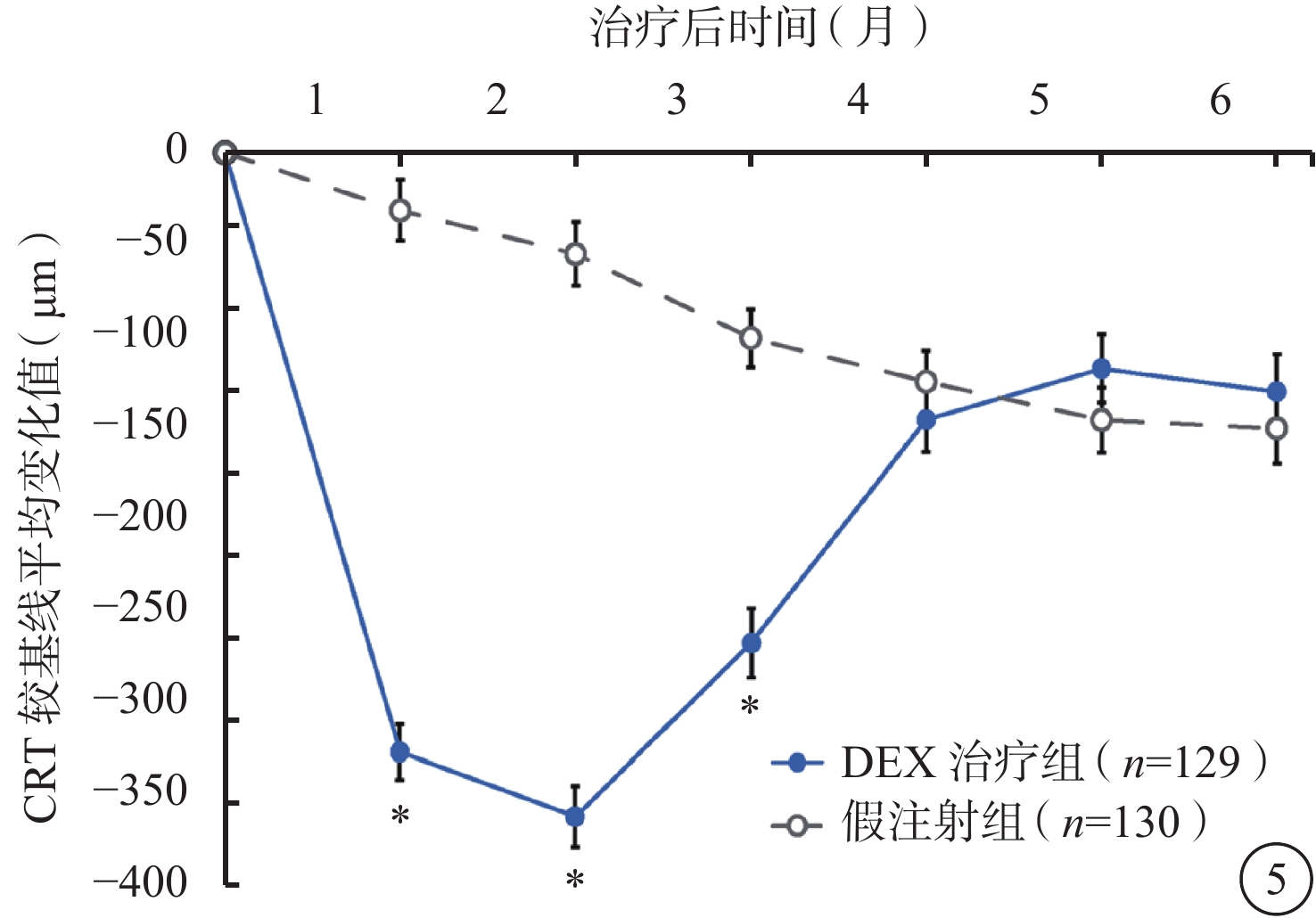 圖5
CRT較基線變化的平均值(mITT人群)。*與假注射組比較,P<0.001
圖5
CRT較基線變化的平均值(mITT人群)。*與假注射組比較,P<0.001
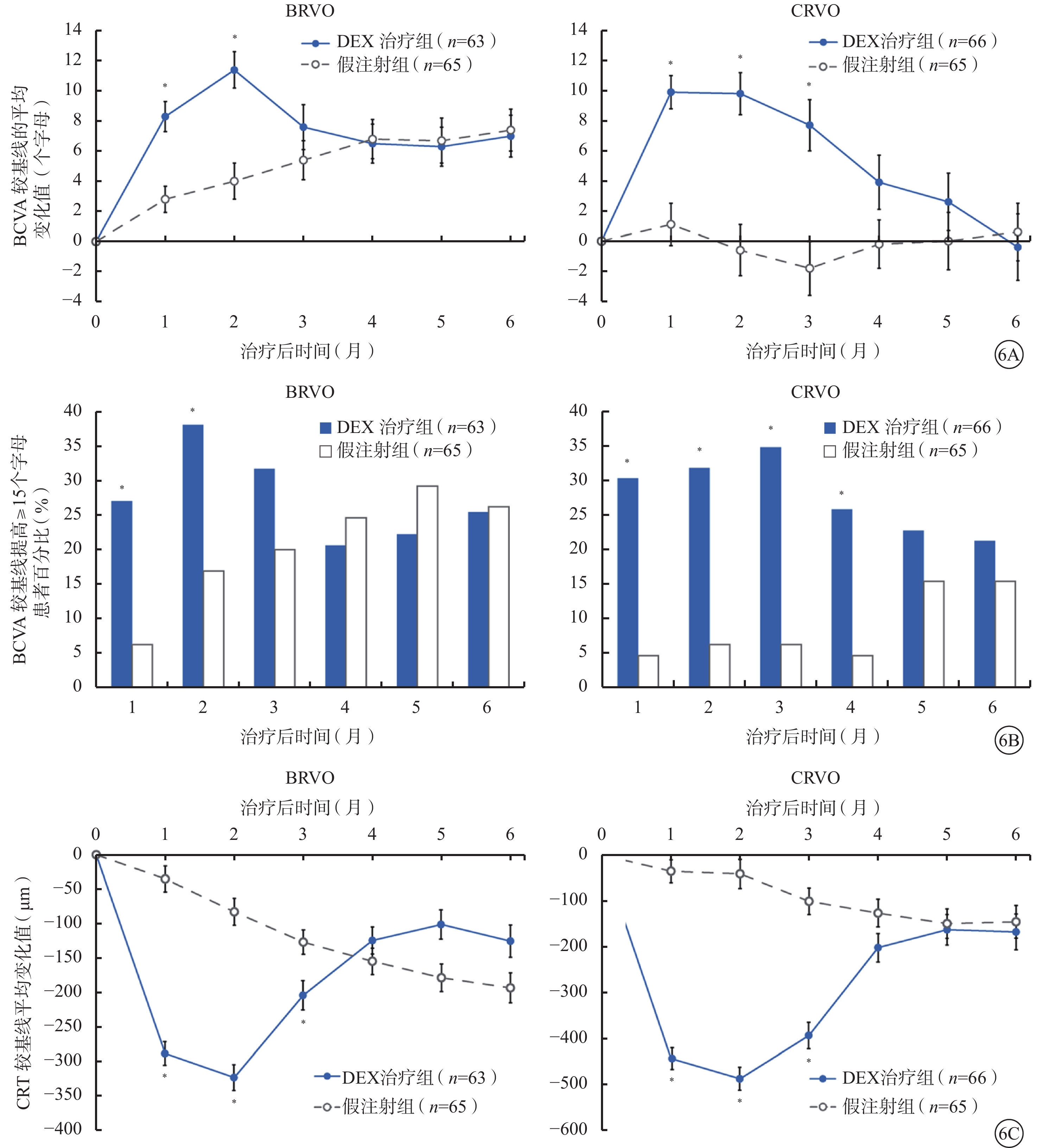
依據RVO類型進行亞組分析,DEX可在治療前3個月改善BRVO、CRVO患者的BCVA和CRT(圖6)。第2個月時,DEX治療組中BRVO、CRVO患者BCVA較基線變化平均值分別為(11.4±9.6)、(9.8±11.0)個字母;假注射組中BRVO、CRVO患者BCVA較基線變化平均值分別為(4.0±10.0)、(?0.6±13.9)個字母。第2個月時,DEX治療組中BRVO、CRVO患者CRT較基線平均降低(323±189)、(487±203)μm;假注射組中BRVO、CRVO患者CRT較基線平均降低(83±187)、(41±256)μm。DEX治療組中BRVO患者在前2~3個月,CRVO患者在前3~4個月期間的療效均顯著優于假注射組(圖6)。無論BRVO還是CRVO患者,基線時ME病程≤90 d者的BCVA預后顯著優于基線時ME病程>90 d者。
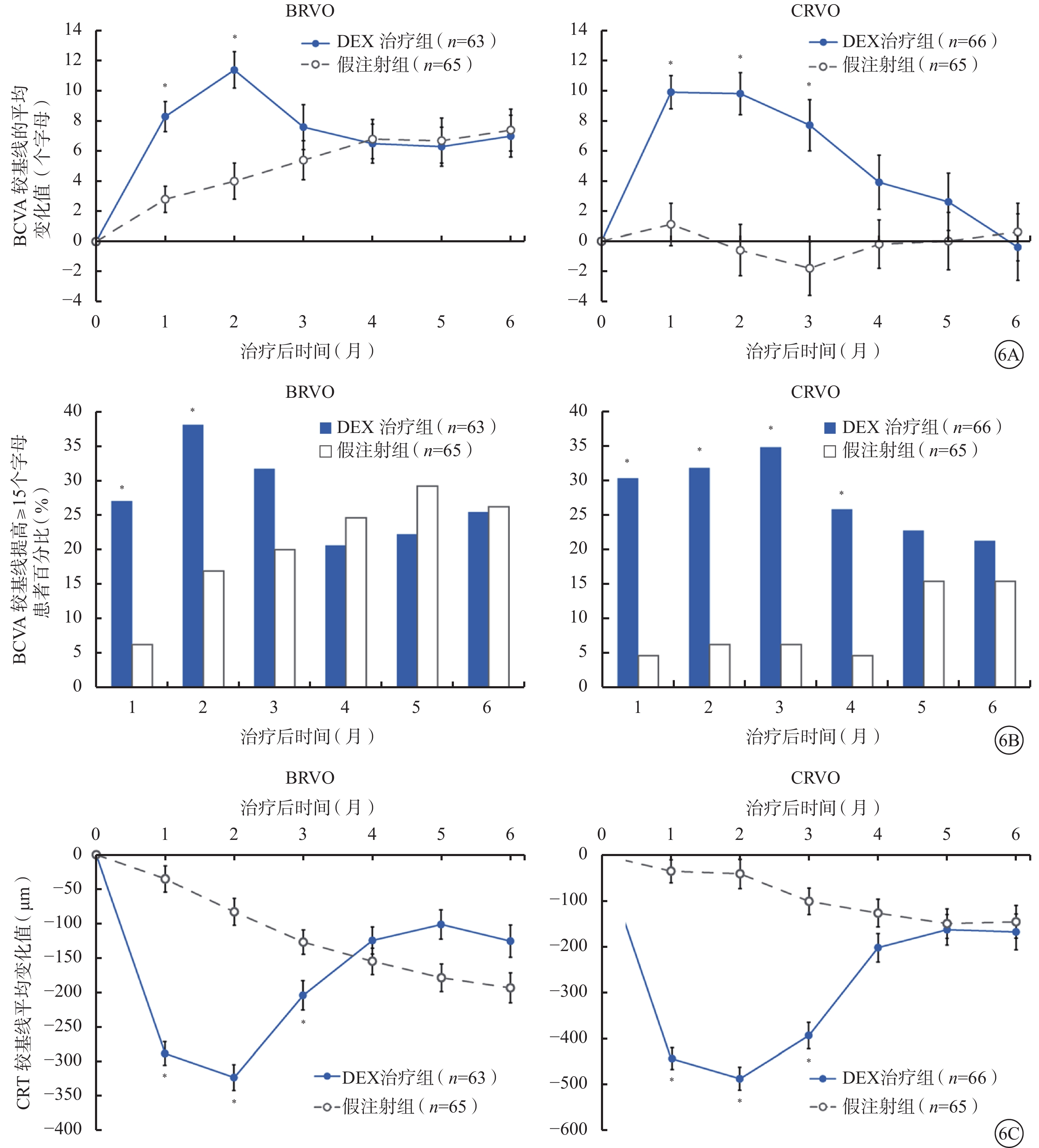 圖6
BRVO和CRVO亞組分析(mITT人群)。6A. BCVA較基線變化的平均值;6B. BCVA較基線提高≥15個字母患者百分比;6C. CRT較基線變化的平均值。與假注射組比較,*P≤0.028
圖6
BRVO和CRVO亞組分析(mITT人群)。6A. BCVA較基線變化的平均值;6B. BCVA較基線提高≥15個字母患者百分比;6C. CRT較基線變化的平均值。與假注射組比較,*P≤0.028
基線訪視時,FFA檢查發現,DEX治療組、假注射組平均視盤新生血管面積分別為(5.26±5.96)、(4.13±3.72)mm2。第6個月時,DEX治療組、假注射組視盤新生血管面積較基線的變化平均值分別為(3.91±7.30)、(2.56±4.58)mm2。
2.2 安全性結果
前6個月中,DEX治療組TEAEs發生率為53.5%(69/129),假注射組TEAEs發生率為31.5%(41/130)。最常見的TEAEs為眼壓升高、結膜出血和結膜充血(表2)。無治療相關的系統性TEAEs發生。在開放標簽的延展研究階段,于第6個月接受DEX再治療的患者TEAEs類型與從基線開始就接受DEX治療的患者TEAEs相同。第二次植入后,并無新的TEAEs發生(表3)。在前6個月中,白內障相關TEAEs在DEX治療組中有2例(1.6%),假注射組為0例。并無患者在研究過程中接受白內障手術治療。
最初的6個月中共有3例患者發生嚴重不良事件(DEX治療組中1例房室傳導阻滯;假注射組中1例玻璃體積血和1例慢性膽囊炎)。在開放標簽的延展研究階段,假注射組中1例患者在接受DEX植入治療后發生嚴重不良事件(腦梗死),但此嚴重不良事件并未認為與治療本身相關。
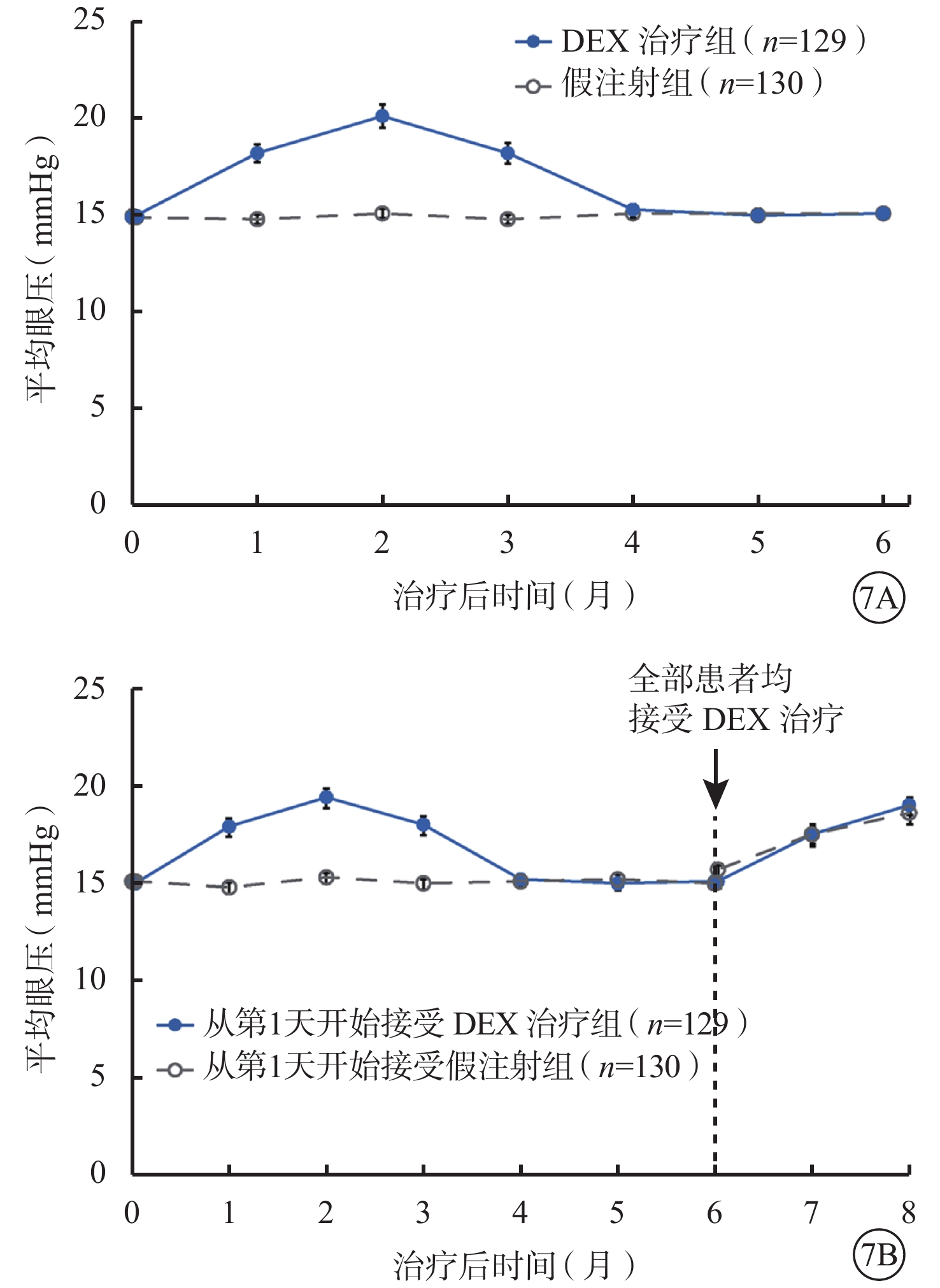
研究的前6個月中,眼壓較基線升高至少10 mmHg者在DEX治療組中為27.1%(35/129),假注射組中為1.5%(2/130);眼壓≥25 mmHg者在DEX治療組中為23.3%(30/129),假注射組中為0%(0/130);眼壓≥35 mmHg者在DEX治療組中為6.2%(8/129),假注射組中為0%(0/130)。DEX治療后平均眼壓峰值出現在第2個月,此后在第4個月時回落至基線水平(圖7A)。在基線和第6個月時均接受了DEX植入治療的患者,平均眼壓的變化在兩次治療過程中相似(圖7B)。
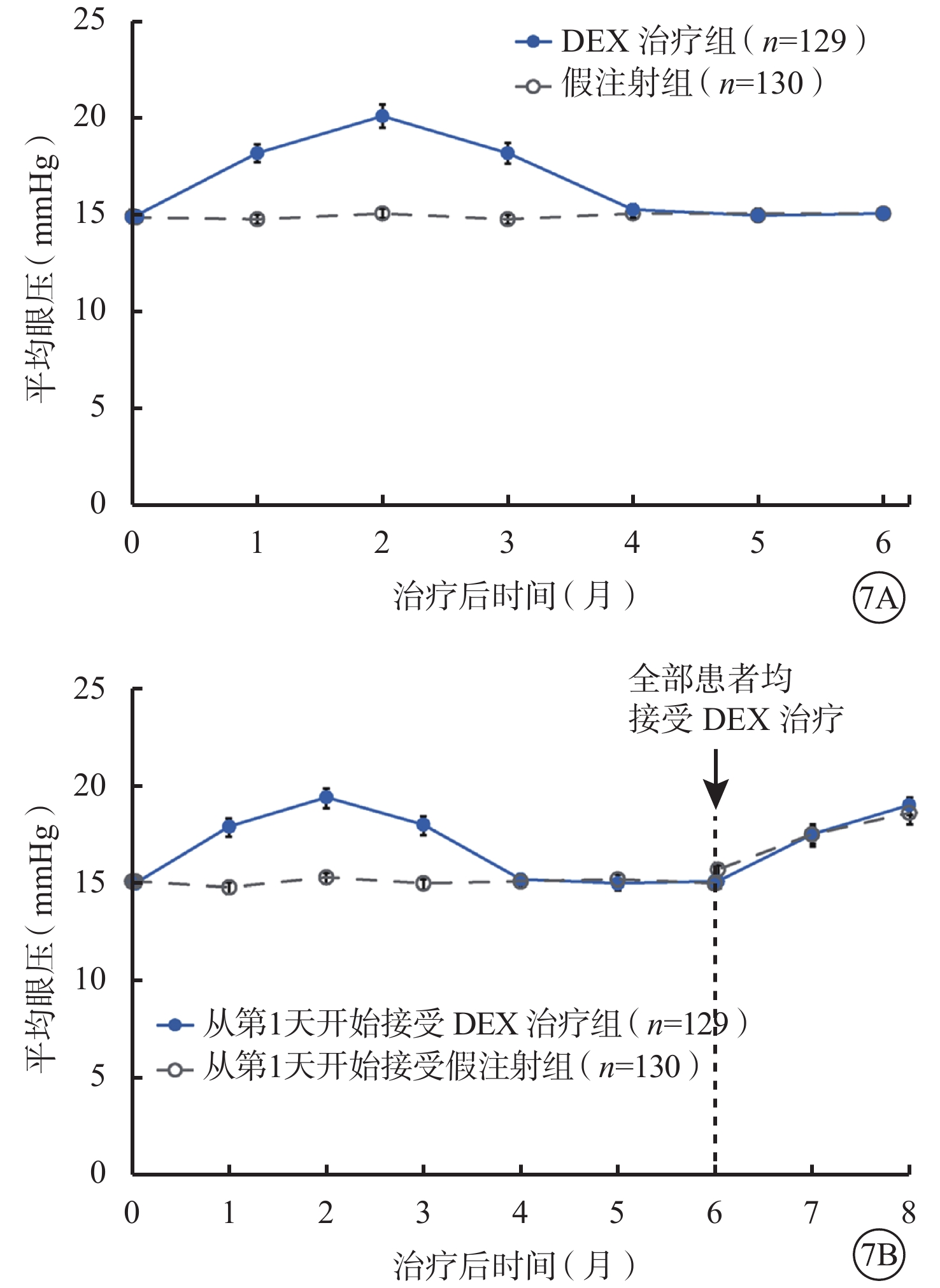 圖7
平均眼壓(安全性人群)。7A. 研究盲態階段基線之后的平均眼壓變化;7B. 在開放標簽階段接受治療的患者在全部8個月內的平均眼壓變化
圖7
平均眼壓(安全性人群)。7A. 研究盲態階段基線之后的平均眼壓變化;7B. 在開放標簽階段接受治療的患者在全部8個月內的平均眼壓變化
對眼壓升高者使用局部降眼壓藥物控制眼壓。在研究進行的8個月中,DEX治療組中34.9%(45/129)的患者和假注射組中13.8%(18/130)的患者(沒有或接受過一次植入治療)使用了降眼壓藥物以控制眼壓。在使用局部降眼壓藥物的患者中,DEX治療組55.6%(24/45)的患者和假注射組83.3%(15/18)的患者只使用了一種藥物。前6個月中,DEX治療組中1例患者因眼壓升高接受激光小梁成形術,假注射組中1例患者因發生青光眼而接受激光周邊虹膜切開術。在開放標簽的延展研究階段,并無患者需要接受降眼壓手術,且整個研究過程中并無患者需要接受抗青光眼手術。
3 討論
本研究結果顯示,DEX 0.7 mg對中國RVO患者有著顯著的臨床療效和良好的耐受性。DEX可快速提高BCVA,并達到本研究的主要研究終點,接受DEX治療的患者平均BCVA提高≥15個字母的時間點早于假注射組。DEX治療組在次要評價指標方面也優于假注射組。DEX治療組在第1、2、3個月時,BCVA較基線變化的平均值、BCVA較基線提高≥15個字母的患者百分比和CRT較基線變化的平均值均顯著優于假注射組。DEX治療組的BCVA在前6個月中變化的平均值也顯著優于假注射組。接受DEX治療的BRVO和CRVO患者均可獲得良好的療效。
DEX在中國患者中的有效性和安全性與全球GENEVA研究中所得結果相近。該研究中,第6個月時,BRVO患者的BCVA較基線變化的平均值優于假注射組,而CRVO組則無差異。本研究結果顯示,第6個月時,CRVO和BRVO患者的BCVA較基線變化的平均值與假注射組均無差異,但CRVO組的療效收益持續時間長于BRVO組,這可能是由于BRVO患者的BCVA和CRT可自發改善所致。目前已知RVO-ME病程越短,則對DEX或抗VEGF藥物治療的反應越好[15-17]。但病程長短難以解釋GENEVA研究和本研究結果的差異,兩項研究基線時ME的病程長度相似(約4~5個月)。第6個月時BRVO患者在兩項研究中結果不同,可能的原因包括患者人群差異、疾病特征或概率因素。
GENEVA全球研究在隨訪的第30、60、90、180天時測定了BCVA,發現其療效峰值為第60天,第180天時DEX 0.7 mg組的BCVA已與假注射組無差異;DEX 0.7 mg組在第90天時CRT較基線變化的平均值顯著優于假注射組,而第180天時差異也已消失[10]。該結果提示DEX治療的有效時長在90~180 d之間,但由于第3個月和第6個月之間并無隨訪訪視點,因此該時長難以具體確定。本研究的隨訪頻率設定為每月一次,進而較GENEVA研究更具優勢,可提供DEX治療對BCVA和CRT的療效隨時間變化和有效時長等額外信息。本研究中DEX治療組和假注射組在第5、6個月時的療效相近,提示對大多數患者再治療間隔為4~5個月。與本研究結果相近,近來一些使用DEX 0.7 mg治療RVO的臨床研究表明再治療平均間隔時間短于6個月[18-20]。如一項美國的回顧性研究(SHASTA研究)表明,DEX 0.7 mg每隔約5個月重復注射治療RVO-ME是有效的[19]。盡管DEX也需每隔4~5個月重復注射方可獲得最佳療效,但注射頻率仍顯著低于抗VEGF藥物玻璃體腔注射。DEX較少的注射次數可減輕治療負擔,也是其治療RVO-ME的優勢。
DEX治療RVO-ME的安全性非常理想且與既往研究結果一致[10, 21]。本研究報道的TEAEs與接受玻璃體腔注射糖皮質激素治療的TEAEs一致,且并無接受DEX治療的患者因TEAEs而退出研究。DEX治療組發生率高于假注射組的常見TEAEs是糖皮質激素相關的眼壓升高和注射相關的結膜充血和出血。眼壓升高可通過局部降眼壓藥物控制,DEX治療組中并無患者因此需要接受抗青光眼手術治療。接受DEX再治療后,患者眼壓升高的平均值與從基線時即接受DEX治療的患者程度相同。此結果與為期3年用DEX治療糖尿病性ME的MEAD研究結果一致,其結果顯示連續DEX治療對眼壓升高程度并無累積效應,且重復治療后眼壓升高的頻率并不增加[22]。由于本研究開放標簽的延展階段只進行了2個月,因此本組患者平均眼壓在研究結束時高于基線水平,與預期一致。MEAD研究中,平均眼壓峰值時間在DEX治療后第1.5~3個月,且在第6個月時降回基線水平[22]。
白內障類TEAEs僅在2例(2/221)有晶狀體眼且接受一次或兩次DEX注射治療的患者中發生,但研究過程中并未因此接受白內障手術治療。本研究的總長度僅為8個月,基線時分入假注射組的患者直到第6個月時才接受DEX治療。既往使用DEX治療RVO的研究表明,在多次DEX注射和治療時間較長時,將很可能發生白內障進展和需要接受白內障手術的情況[6, 23]。
本研究的局限性在于預定的再治療時間間隔為6個月。這雖然有助于我們評估單次DEX植入的療效持續時長,但與間隔4~5個月這一較短的再治療方案相比,BCVA和CRT預后并非最優結果。
本研究結果表明,DEX 0.7 mg可顯著提高中國RVO-ME患者的視力和解剖預后。單次DEX治療后的前3~4個月內,BCVA提高和CRT改善均顯著優于假注射組。為獲得最佳療效,再治療時間間隔須短于6個月。
視網膜靜脈阻塞(RVO)是一種常見的危害視功能的視網膜疾病。在基于人群的北京眼病研究中,大北京地區40歲及以上成年人中RVO患病率約為0.7%,且隨年齡增長而增加[1]。視網膜分支靜脈阻塞(BRVO)和視網膜中央靜脈阻塞(CRVO)是最主要兩種類型,黃斑水腫(ME)是危害其視功能的主要原因[2]。目前治療RVO-ME的方法主要有玻璃體腔注射抗血管內皮生長因子(VEGF)藥物、糖皮質激素,以及BRVO格柵樣激光光凝治療。抗VEGF藥物治療對RVO-ME通常有效,但需頻繁注射且并非對所有患者有效[3]。糖皮質激素治療RVO符合發病機制[4],但主要副作用是糖皮質激素相關眼壓升高和白內障加重[5, 6]。
地塞米松玻璃體腔植入劑(DEX)是一種由NOVADUR@固態聚合物包裹的,內含糖皮質激素地塞米松眼內植入物,可持續緩慢釋放藥物且可生物降解[7]。該植入物通過22G一次性給藥器經睫狀體扁平部植入玻璃體腔[8]。靈長類動物實驗結果證實,玻璃體腔DEX植入后前2個月地塞米松濃度均維持較高水平,此后逐漸下降,且在前6個月中均可檢測到[9]。全球GENEVA研究對比觀察了DEX 0.7 mg、DEX 0.35 mg和假注射治療BRVO、CRVO繼發ME的療效,DEX 0.7 mg和0.35mg較假注射均可顯著改善黃斑中心凹視網膜厚度(CRT),并在單次植入后前3個月患者最佳矯正視力(BCVA)得到提高[6, 10]。DEX 0.7 mg治療組在治療后1周時較假注射組更可獲得3行以上的視力提高[11]。
基于GENEVA研究結果,美國和歐洲批準DEX 0.7 mg用于RVO-ME的治療。但GENEVA研究中DEX 0.7 mg治療組僅納入38例亞裔患者,且DEX 0.7 mg治療RVO-ME的安全性和有效性尚未在亞洲人群得到充分證實[10]。由于中國大陸40歲及以上成年人中原發性開角型青光眼和原發性閉角型青光眼患病率分別約為0.7%和1.4%[12],因此在中國人群中評估糖皮質激素相關眼壓升高發生率和轉歸以明確DEX的安全性顯得尤為重要。本研究旨在評估DEX 0.7 mg較假注射對照治療BRVO、CRVO繼發ME在中國患者中的安全性和有效性。
1 對象和方法
1.1 研究方法
隨機、雙盲、假注射對照、多中心(13個中心)為期6個月的3期臨床研究,此后接續為期2個月的開放標簽的延展研究,以評估DEX 0.7 mg治療RVO-ME的療效。DEX 0.7 mg單次注射的安全性和有效性在本研究的隨機階段以假注射為對照統計得出;DEX 0.7 mg的安全性進一步在后續開放標簽的延展研究中進行評估,此階段全部患者均接受DEX 0.7 mg植入治療。本研究于2012年9月至2014年5月依據中國藥物臨床試驗管理規范法規和準則在中國開展。本研究開始時,中國國內尚無已經批準上市的用于治療RVO-ME的抗VEGF藥物。本研究設計與GENEVA全球研究類似,評估DEX 0.7 mg對中國患者的療效;研究方案經各中心獨立的倫理委員會批準通過,全部患者均簽署書面知情同意書。本研究注冊于www.ClinicalTrials.gov,注冊號為NCT01660802。
1.2 患者選擇
患者選擇評估包括篩選訪視(第?14~?1天)和基線訪視(第1天)。年齡≥18歲;熒光素眼底血管造影(FFA)檢查評估為非缺血型BRVO或CRVO;光相干斷層掃描(OCT)檢查可見黃斑區視網膜增厚并累及中心凹;BRVO、CRVO繼發ME病程分別在篩選訪視前6~12個月和6~9個月;患者視力下降與ME相關;BCVA≥34個字母且≤68個字母(對應的Snellen視力分別為20/200和20/50)[13]。BCVA檢查采用早期治療糖尿病視網膜病變研究組視力表進行。OCT檢查采用德國Heidelberg公司Spectralis OCT儀,CRT≥320 μm,或德國Zeiss公司Cirrus OCT儀,CRT≥300 μm。篩選訪視時觀察黃斑中心凹1 mm范圍內CRT厚度。
主要排除標準:缺血型RVO,定義為FFA上無灌注區累及黃斑中心凹且超過10個視盤面積;有青光眼病史;基線前3個月內接受過玻璃體腔注射抗VEGF藥物或糖皮質激素治療;基線前3個月內接受過激光光凝或內眼手術治療;經研究者評估,患者存在影響BCVA提高15個字母以上或可能影響ME或BCVA的其他眼部情況,如嚴重黃斑缺血、黃斑前膜或黃斑中心凹萎縮;篩選訪視時患者屈光間質混濁影響臨床或眼底彩色照相評估,包括但不限于視網膜前或玻璃體積血、晶狀體混濁;致密的黃斑區積血且眼底紅光反射消失。其他重要排除標準:既往接受過經平坦部玻璃體切割手術;任一眼存在活動性細菌、病毒、寄生蟲或真菌感染;任一眼曾因接受糖皮質激素治療而發生眼壓升高;基線前1個月內曾接受糖皮質激素口服、靜脈、肌內注射、硬膜外、直腸內或皮膚外用治療;基線前3個月內曾接受免疫抑制劑、免疫調節劑、抗代謝藥和(或)氮芥類藥物治療;基線前2周曾接受眼局部糖皮質激素點眼或中藥治療;對側眼BCVA<34個字母;基線訪視BCVA較篩選訪視提高>10個字母;經研究者評估可能影響研究結果的其他因素。
1.3 治療
基線訪視時(第1天),患者按1:1隨機分為DEX治療組、假注射組,分別接受DEX 0.7 mg或假注射治療。治療組再分別按RVO類型分為BRVO或CRVO亞組。基線訪視評估后接受研究方案的相應治療。DEX治療組,DEX 0.7 mg由一次性給藥器注射植入玻璃體腔[8];假注射組,注射過程使用不帶針頭的給藥器按壓結膜。
患者在治療后第6個月時接受評估判斷是否可進入第二階段研究。若患者BCVA<84個字母(約相當于Snellen視力20/20),仍殘余ME(CRT>250 μm、視網膜內囊腔及黃斑中心區域內外視網膜厚度增加),患者可接受再次治療,且研究者評估再治療過程不會把患者置于重大風險之中。再治療時DEX治療組和假注射組均使用DEX 0.7 mg。
挽救治療。治療≥3個月后,若患者BCVA在連續兩次間隔4周的復診時,因RVO-ME加重而導致視力較基線下降≥10個字母,行挽救性激光光凝治療。任何情況下,若研究者認為挽救性激光光凝對患者有益時均可給予挽救治療。接受挽救性激光光凝治療的患者仍保留在研究中且可從第6個月開始接受開放標簽的DEX注射治療。
1.4 隨訪和評估
治療后1~8個月,每月隨訪一次。安全性評估由研究者在治療和再治療后第1天時進行。每月隨訪時均進行有效性評估,包括BCVA和OCT。篩選訪視和第6個月時對FFA檢查所見熒光素滲漏程度進行評估。讀片中心處于盲態的讀片師對FFA所示新生血管是否存在和面積大小通過對照標準圖片進行判讀。每次訪視時的主要安全性評估內容包括裂隙燈顯微鏡、Goldmann壓平眼壓計、OCT檢查等以及治療相關不良事件(TEAEs)的發生情況。研究者使用裂隙燈顯微鏡,對照年齡相關眼部疾病研究臨床晶狀體分級系統的標準圖片,對患者晶狀體核、皮質和后囊下混濁存在與否及嚴重程度進行分級[14]。TEAEs定義為基線之后發生或嚴重程度加重的不良事件,及任何嚴重不良事件。全部患者、對患者每月進行隨訪評估的研究者、收集有效數據的研究人員和讀片中心(美國Doheny圖像閱讀中心)對OCT圖像進行評估的讀片師均對患者的分組情況處于盲態。
主要有效終點為治療后6個月中患者BCVA較基線提高≥15個字母所需的時間。關鍵次要終點為治療后6個月每次隨訪時BCVA較基線提高≥15個字母的患者百分比,以及OCT檢查所示CRT較基線的變化。治療后6個月內患者BCVA較基線的變化值通過曲線下面積(AUC)的方式進行評估。依據RVO診斷(BRVO或CRVO)的亞組分析也作為重要評價指標。
1.5 數據分析和統計方法
由全部接受隨機分組和治療的患者組成的修正治療人群(mITT)用于主要有效終點的分析。全部遵守和無嚴重違反研究規程且接受隨機分組和治療的患者均用于主要終點的輔助性分析。依據患者真實接受的治療,對由全部接受治療的患者組成的安全性人群的數據進行評估得出安全性數據。
采用SAS 9.3軟件進行統計分析,雙側檢驗α=0.05。接受挽救治療之后的患者數據不用于任何有效性分析,且挽救治療之后的有效性數據均設置為缺失,用末次觀察轉結法代替。
采用Kaplan-Meier生存曲線法分析治療后前6個月患者BCVA較基線提高≥15個字母所需的時間。其中,治療后6個月無反應的患者將從第6個月時被剔除;若患者在前6個月內退出研究,則該時間點設定為末次接受視力測定的時間。BCVA提高≥15個字母之前接受挽救治療的患者也在接受初次挽救治療的時間點被剔除。治療組間的累積有效率用秩和檢驗進行比較。
治療后6個月內平均BCVA通過AUC方法計算得出。AUC通過梯形面積疊加法,以BCVA和測量時的研究天數計算得出。DEX治療組、假注射組患者之間平均BCVA比較行雙因素方差分析(ANOVA),其中治療分組和RVO類型(BRVO或CRVO)均被設定為固定因素。其他全部關于BCVA和OCT數據的次要分析,連續變量使用ANOVA法,分類變量使用Mantel-Haenszel法依據RVO類型(BRVO或CRVO)分層分析。TEAEs按照MedDRA第17.0版推薦用詞編碼,與白內障相關的全部TEAEs(白內障、糖尿病性白內障、核性白內障、囊下性白內障、皮質性白內障或晶狀體核混濁)均被評估。
預計每組樣本量為130例患者時,BCVA在治療后前6個月較基線提高≥15個字母的時間兩組存在差異的效力為85%,假定假注射組的累積反應為22.5%,DEX組相對假注射組的固定風險比為2。
2 結果
共328例患者接受篩選訪視,其中262例患者進入研究并隨機分組接受DEX或假注射治療。納入研究并隨機分組的262例患者中,因未接受治療而從mITT人群中剔除3例(DEX治療組1例、假注射組2例)。因此,共計259例患者259只眼納入最終分析(表1)。所有患者均為亞裔。DEX治療組男性患者所占比例(53.5%)較假注射組(41.5%)更大,但兩組患眼疾病特征無差異。259只眼中,255只眼(98.5%)為有晶狀體眼;BRVO、CRVO約各占一半。DEX治療組、假注射組前6個月雙盲階段研究的完成率分別為97.7%、94.6%。假注射組中1例患者因不良事件而退出研究,為黃斑囊樣水腫者(圖1)。
圖1
本研究患者入組流程圖(mITT人群)
第6個月時,共203例患者進入后續開放標簽的DEX注射延展治療階段,包括DEX治療組107例(BRVO 53例、CRVO 54例)及假注射組96例(BRVO 41例、CRVO 55例)。249例完成前段研究的患者中,有46例患者(BRVO 30例、CRVO 16例)在第6個月時不再接受治療;其中,25例(54.3%)因CRT≤250 μm而不再需要治療。DEX治療組中100%(107/107)的患者均接受了再次DEX注射治療;而假注射組中99%(95/96)的患者在第6個月時接受DEX注射治療并完成后續為期2個月的開放標簽延展研究。
DEX治療組(BRVO、CRVO各7例)中10.9%(14/129)的患者和假注射組(BRVO 9例、CRVO 2例)中8.5%(11/130)的患者接受視網膜激光光凝挽救治療。DEX治療組中71.4%(10/14)的患者和假注射組中54.5%(6/11)的患者初次激光治療時間為第6個月。
2.1 有效性結果
DEX可快速提高BCVA。生存曲線分析結果顯示,DEX治療組BCVA提高≥15個字母所需時間顯著短于假注射組(P<0.001)(圖2)。DEX治療組和假注射組累積有效曲線從第1次隨訪(第1個月)時即開始分離并持續至第6個月。經Cox回歸模型依據基線時RVO類型、年齡和性別調整后對全部完成隨訪的患者進行分析,證實DEX治療組和假注射組總體治療有效率存在顯著差異(P<0.001)。從該模型計算出的估計風險比為2.4(95%可信區間1.6~3.7),即DEX治療組患者獲得視力提高≥15個字母的概率是假注射組的2.4倍。
圖2
BCVA較基線提高≥15個字母所需時間的Kaplan-Meier分析圖(mITT人群)。*與假注射組比較,P<0.001
DEX治療組中BCVA較基線的變化值和BCVA較基線提高≥15個字母的患者百分比在第1、2、3個月時均顯著高于假注射組(P<0.001)(圖3,4)。第2個月時(效應峰值),DEX治療組BCVA較基線平均提高(10.6±10.4)個字母,假注射組BCVA較基線平均提高(1.7±12.3)個字母(P<0.001);DEX治療組、假注射組BCVA較基線提高≥15個字母患者百分比分別為34.9%、11.5%(P<0.001)。DEX治療組、假注射組前6個月BCVA較基線變化平均值為(6.7±9.0)、(2.5±10.0)個字母(P<0.001)。
圖3
BCVA較基線的平均變化值(mITT人群)。*與假注射組比較,P<0.001
圖4
BCVA較基線提高≥15個字母患者百分比(mITT人群)。*與假注射組比較,P<0.001
DEX治療組的解剖預后優于假注射組。第1、2、3個月時,DEX治療組CRT較基線的平均下降值顯著高于假注射組(P<0.001)(圖5)。第2個月時(效應峰值),DEX治療組CRT較基線平均降低(407±212) μm,假注射組CRT較基線平均降低(62±224)μm(P<0.001)。
圖5
CRT較基線變化的平均值(mITT人群)。*與假注射組比較,P<0.001
依據RVO類型進行亞組分析,DEX可在治療前3個月改善BRVO、CRVO患者的BCVA和CRT(圖6)。第2個月時,DEX治療組中BRVO、CRVO患者BCVA較基線變化平均值分別為(11.4±9.6)、(9.8±11.0)個字母;假注射組中BRVO、CRVO患者BCVA較基線變化平均值分別為(4.0±10.0)、(?0.6±13.9)個字母。第2個月時,DEX治療組中BRVO、CRVO患者CRT較基線平均降低(323±189)、(487±203)μm;假注射組中BRVO、CRVO患者CRT較基線平均降低(83±187)、(41±256)μm。DEX治療組中BRVO患者在前2~3個月,CRVO患者在前3~4個月期間的療效均顯著優于假注射組(圖6)。無論BRVO還是CRVO患者,基線時ME病程≤90 d者的BCVA預后顯著優于基線時ME病程>90 d者。
圖6
BRVO和CRVO亞組分析(mITT人群)。6A. BCVA較基線變化的平均值;6B. BCVA較基線提高≥15個字母患者百分比;6C. CRT較基線變化的平均值。與假注射組比較,*P≤0.028
基線訪視時,FFA檢查發現,DEX治療組、假注射組平均視盤新生血管面積分別為(5.26±5.96)、(4.13±3.72)mm2。第6個月時,DEX治療組、假注射組視盤新生血管面積較基線的變化平均值分別為(3.91±7.30)、(2.56±4.58)mm2。
2.2 安全性結果
前6個月中,DEX治療組TEAEs發生率為53.5%(69/129),假注射組TEAEs發生率為31.5%(41/130)。最常見的TEAEs為眼壓升高、結膜出血和結膜充血(表2)。無治療相關的系統性TEAEs發生。在開放標簽的延展研究階段,于第6個月接受DEX再治療的患者TEAEs類型與從基線開始就接受DEX治療的患者TEAEs相同。第二次植入后,并無新的TEAEs發生(表3)。在前6個月中,白內障相關TEAEs在DEX治療組中有2例(1.6%),假注射組為0例。并無患者在研究過程中接受白內障手術治療。
最初的6個月中共有3例患者發生嚴重不良事件(DEX治療組中1例房室傳導阻滯;假注射組中1例玻璃體積血和1例慢性膽囊炎)。在開放標簽的延展研究階段,假注射組中1例患者在接受DEX植入治療后發生嚴重不良事件(腦梗死),但此嚴重不良事件并未認為與治療本身相關。
研究的前6個月中,眼壓較基線升高至少10 mmHg者在DEX治療組中為27.1%(35/129),假注射組中為1.5%(2/130);眼壓≥25 mmHg者在DEX治療組中為23.3%(30/129),假注射組中為0%(0/130);眼壓≥35 mmHg者在DEX治療組中為6.2%(8/129),假注射組中為0%(0/130)。DEX治療后平均眼壓峰值出現在第2個月,此后在第4個月時回落至基線水平(圖7A)。在基線和第6個月時均接受了DEX植入治療的患者,平均眼壓的變化在兩次治療過程中相似(圖7B)。
圖7
平均眼壓(安全性人群)。7A. 研究盲態階段基線之后的平均眼壓變化;7B. 在開放標簽階段接受治療的患者在全部8個月內的平均眼壓變化
對眼壓升高者使用局部降眼壓藥物控制眼壓。在研究進行的8個月中,DEX治療組中34.9%(45/129)的患者和假注射組中13.8%(18/130)的患者(沒有或接受過一次植入治療)使用了降眼壓藥物以控制眼壓。在使用局部降眼壓藥物的患者中,DEX治療組55.6%(24/45)的患者和假注射組83.3%(15/18)的患者只使用了一種藥物。前6個月中,DEX治療組中1例患者因眼壓升高接受激光小梁成形術,假注射組中1例患者因發生青光眼而接受激光周邊虹膜切開術。在開放標簽的延展研究階段,并無患者需要接受降眼壓手術,且整個研究過程中并無患者需要接受抗青光眼手術。
3 討論
本研究結果顯示,DEX 0.7 mg對中國RVO患者有著顯著的臨床療效和良好的耐受性。DEX可快速提高BCVA,并達到本研究的主要研究終點,接受DEX治療的患者平均BCVA提高≥15個字母的時間點早于假注射組。DEX治療組在次要評價指標方面也優于假注射組。DEX治療組在第1、2、3個月時,BCVA較基線變化的平均值、BCVA較基線提高≥15個字母的患者百分比和CRT較基線變化的平均值均顯著優于假注射組。DEX治療組的BCVA在前6個月中變化的平均值也顯著優于假注射組。接受DEX治療的BRVO和CRVO患者均可獲得良好的療效。
DEX在中國患者中的有效性和安全性與全球GENEVA研究中所得結果相近。該研究中,第6個月時,BRVO患者的BCVA較基線變化的平均值優于假注射組,而CRVO組則無差異。本研究結果顯示,第6個月時,CRVO和BRVO患者的BCVA較基線變化的平均值與假注射組均無差異,但CRVO組的療效收益持續時間長于BRVO組,這可能是由于BRVO患者的BCVA和CRT可自發改善所致。目前已知RVO-ME病程越短,則對DEX或抗VEGF藥物治療的反應越好[15-17]。但病程長短難以解釋GENEVA研究和本研究結果的差異,兩項研究基線時ME的病程長度相似(約4~5個月)。第6個月時BRVO患者在兩項研究中結果不同,可能的原因包括患者人群差異、疾病特征或概率因素。
GENEVA全球研究在隨訪的第30、60、90、180天時測定了BCVA,發現其療效峰值為第60天,第180天時DEX 0.7 mg組的BCVA已與假注射組無差異;DEX 0.7 mg組在第90天時CRT較基線變化的平均值顯著優于假注射組,而第180天時差異也已消失[10]。該結果提示DEX治療的有效時長在90~180 d之間,但由于第3個月和第6個月之間并無隨訪訪視點,因此該時長難以具體確定。本研究的隨訪頻率設定為每月一次,進而較GENEVA研究更具優勢,可提供DEX治療對BCVA和CRT的療效隨時間變化和有效時長等額外信息。本研究中DEX治療組和假注射組在第5、6個月時的療效相近,提示對大多數患者再治療間隔為4~5個月。與本研究結果相近,近來一些使用DEX 0.7 mg治療RVO的臨床研究表明再治療平均間隔時間短于6個月[18-20]。如一項美國的回顧性研究(SHASTA研究)表明,DEX 0.7 mg每隔約5個月重復注射治療RVO-ME是有效的[19]。盡管DEX也需每隔4~5個月重復注射方可獲得最佳療效,但注射頻率仍顯著低于抗VEGF藥物玻璃體腔注射。DEX較少的注射次數可減輕治療負擔,也是其治療RVO-ME的優勢。
DEX治療RVO-ME的安全性非常理想且與既往研究結果一致[10, 21]。本研究報道的TEAEs與接受玻璃體腔注射糖皮質激素治療的TEAEs一致,且并無接受DEX治療的患者因TEAEs而退出研究。DEX治療組發生率高于假注射組的常見TEAEs是糖皮質激素相關的眼壓升高和注射相關的結膜充血和出血。眼壓升高可通過局部降眼壓藥物控制,DEX治療組中并無患者因此需要接受抗青光眼手術治療。接受DEX再治療后,患者眼壓升高的平均值與從基線時即接受DEX治療的患者程度相同。此結果與為期3年用DEX治療糖尿病性ME的MEAD研究結果一致,其結果顯示連續DEX治療對眼壓升高程度并無累積效應,且重復治療后眼壓升高的頻率并不增加[22]。由于本研究開放標簽的延展階段只進行了2個月,因此本組患者平均眼壓在研究結束時高于基線水平,與預期一致。MEAD研究中,平均眼壓峰值時間在DEX治療后第1.5~3個月,且在第6個月時降回基線水平[22]。
白內障類TEAEs僅在2例(2/221)有晶狀體眼且接受一次或兩次DEX注射治療的患者中發生,但研究過程中并未因此接受白內障手術治療。本研究的總長度僅為8個月,基線時分入假注射組的患者直到第6個月時才接受DEX治療。既往使用DEX治療RVO的研究表明,在多次DEX注射和治療時間較長時,將很可能發生白內障進展和需要接受白內障手術的情況[6, 23]。
本研究的局限性在于預定的再治療時間間隔為6個月。這雖然有助于我們評估單次DEX植入的療效持續時長,但與間隔4~5個月這一較短的再治療方案相比,BCVA和CRT預后并非最優結果。
本研究結果表明,DEX 0.7 mg可顯著提高中國RVO-ME患者的視力和解剖預后。單次DEX治療后的前3~4個月內,BCVA提高和CRT改善均顯著優于假注射組。為獲得最佳療效,再治療時間間隔須短于6個月。