引用本文: 楊洋, 楊波, 柳小麗, 佟柏楠, 肖駿. Crouzon綜合征一例. 中華眼底病雜志, 2018, 34(1): 76-77. doi: 10.3760/cma.j.issn.1005-1015.2018.01.022 復制
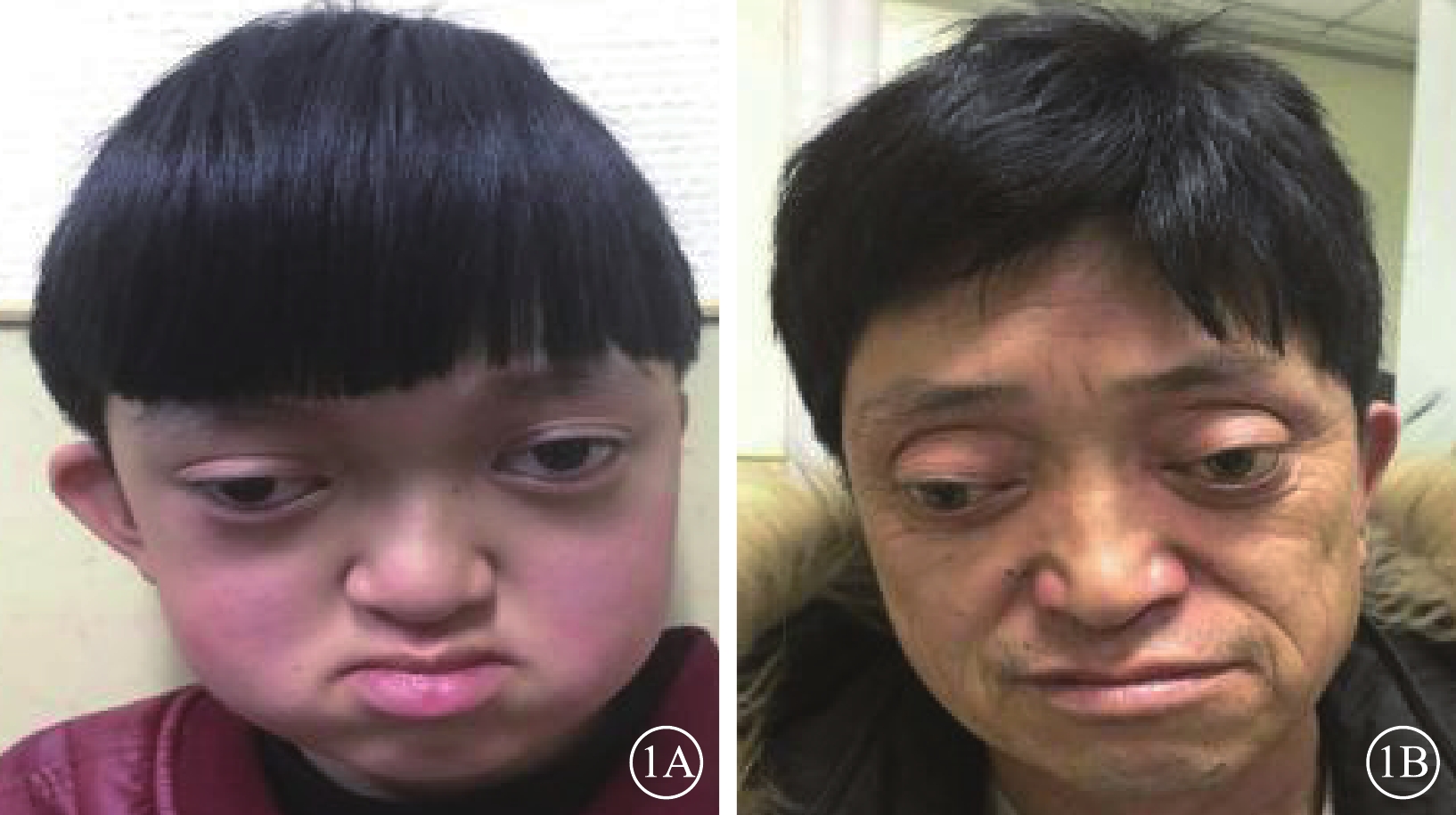
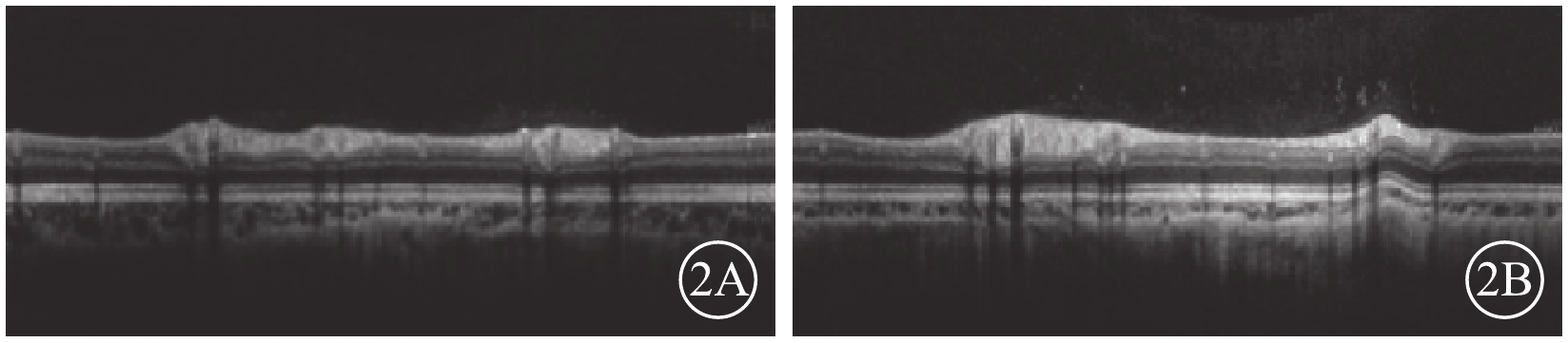
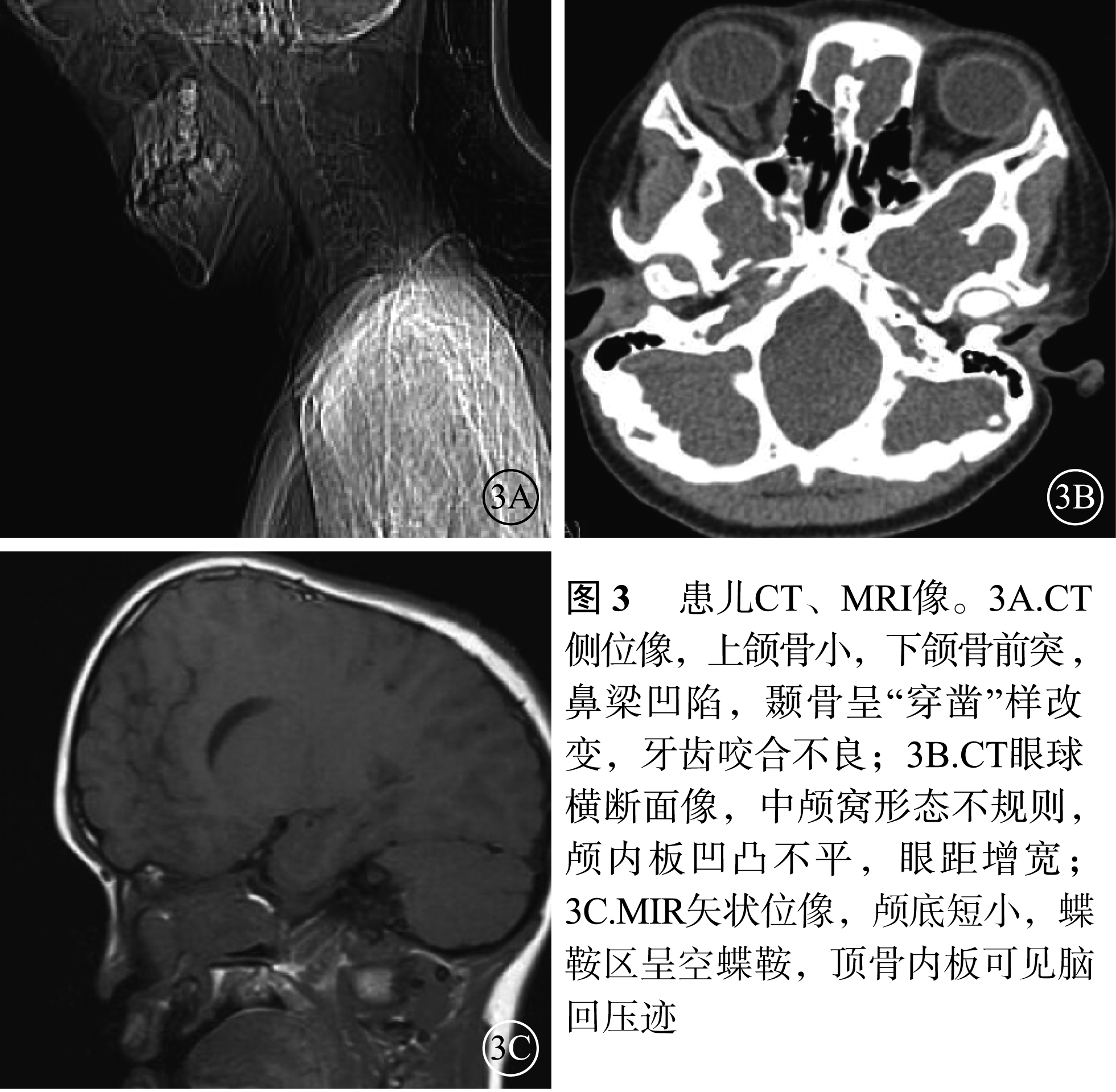
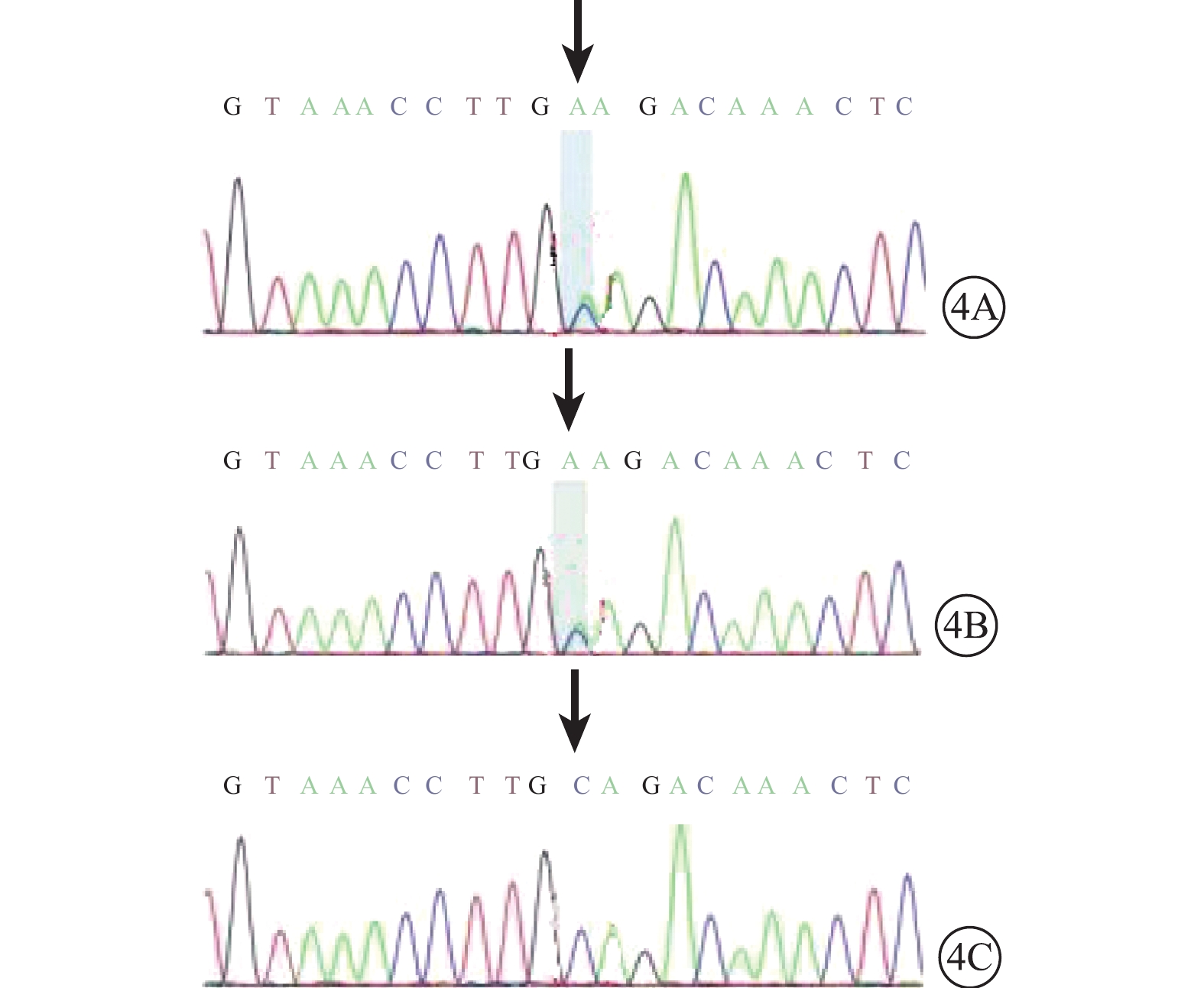
患兒男,12歲。因右眼視力下降2個月于2017年1月9日來我院眼科就診。患兒出生時即有雙眼突出,眼距較寬;智力發育正常。全身檢查:頭顱畸形、前額突出、鼻梁凹陷呈鸚鵡嘴狀鼻(圖1A);其他全身檢查未見異常。家族中其父親與患兒有相同特殊面容(圖1B);否認家族中有其他類似病例。眼部檢查:右眼視力0.08,手光/?1.00×45→0.16;左眼視力0.3,?1.75/?1.50×90→0.9。右眼眼壓15 mmHg(1 mmHg=0.133 kPa),左眼眼壓17 mmHg。雙眼眼位正;眼球突出度均為23 mm,眶距105 mm。輕度眼球震顫。雙眼眼前節檢查正常;瞳孔直接、間接對光反射正常。雙眼視盤邊界清楚,顏色略淡;視網膜血管走形、黃斑中心凹反光正常。光相干斷層掃描(OCT)檢查,雙眼視盤顳側神經纖維層(RNFL)厚度變薄(圖2A,2B)。雙眼視野、視覺誘發電位、色覺檢查均未見異常。耳聲阻抗檢查,左耳B形曲線,中耳積液;右耳正常。聽力檢查,左耳骨傳導下降;右耳正常。視神經孔、眼眶及頭部CT檢查,上頜骨小,下頜骨前突,鼻梁骨凹陷(圖3A);顱中窩形態不規則,眼距增寬,眼球略突出(圖3B);雙側視神經孔略增粗。頭部MRI檢查,左側放射冠區脫髓鞘;雙側篩竇及上頜竇炎;小腦扁桃體下疝畸形(圖3C)。其父親右眼視力1.0,左眼視力0.6。眼前節、眼底、OCT檢查結果均正常。基因檢查,患兒、其父親均發現耳聾相關基因成纖維細胞生長因子受體2(FGFR2)基因存在c.833G>T chr10:123279599 p.Cys278Phe雜合突變(圖4A,4B);其母親檢查結果正常(圖4C)。家系驗證結果顯示該突變來自患兒父親。線粒體基因組基因檢測,線粒體基因相關熱點突變未發現明確異常。診斷:Crouzon綜合征。
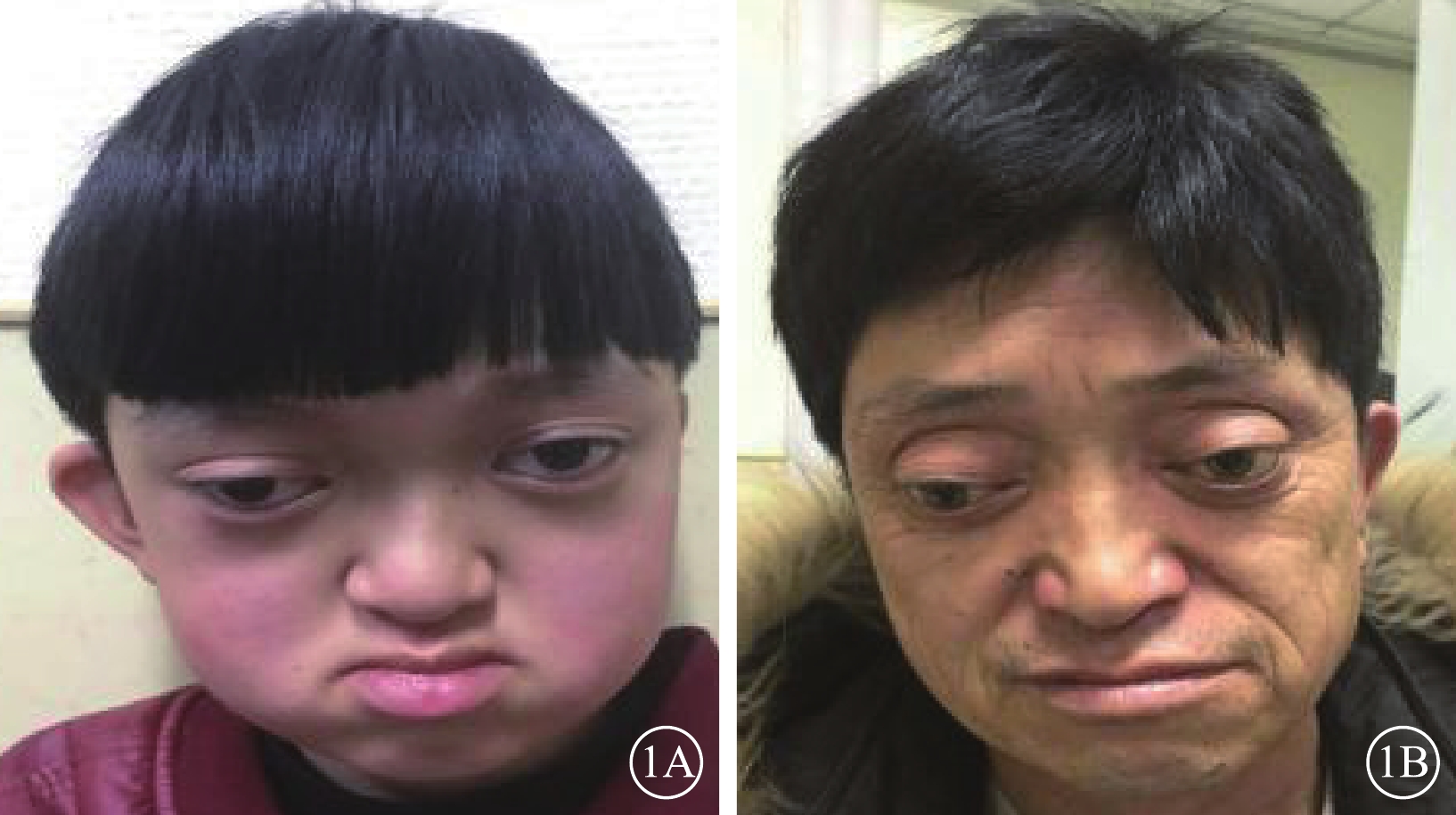 圖1
患兒和其父親面貌正面彩色像。1A. 患兒;1B. 父親。眼距增寬,眼球突出,鼻根低平
圖1
患兒和其父親面貌正面彩色像。1A. 患兒;1B. 父親。眼距增寬,眼球突出,鼻根低平
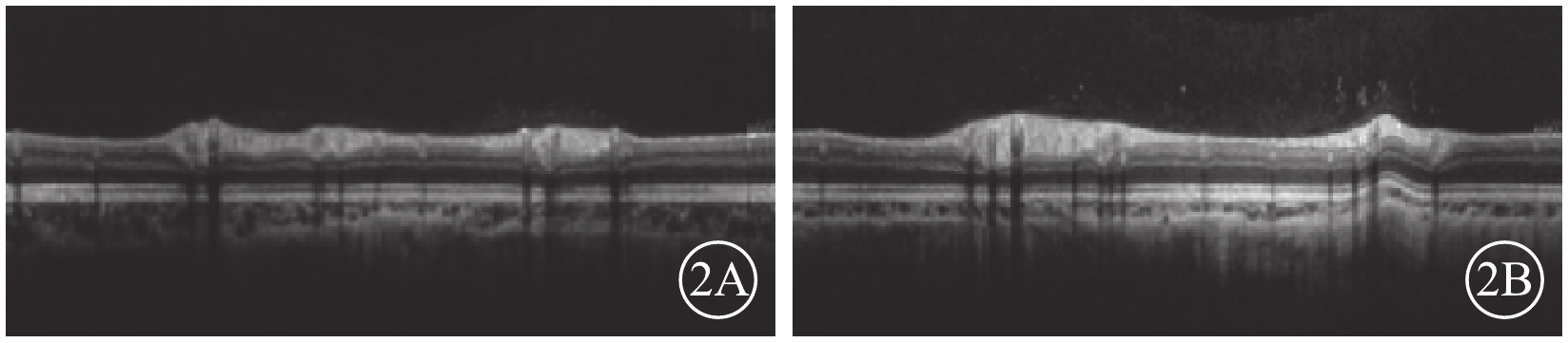 圖2
患兒雙眼視盤OCT像。2A. 右眼;2B. 左眼。顳側RNFL厚度變薄
圖2
患兒雙眼視盤OCT像。2A. 右眼;2B. 左眼。顳側RNFL厚度變薄
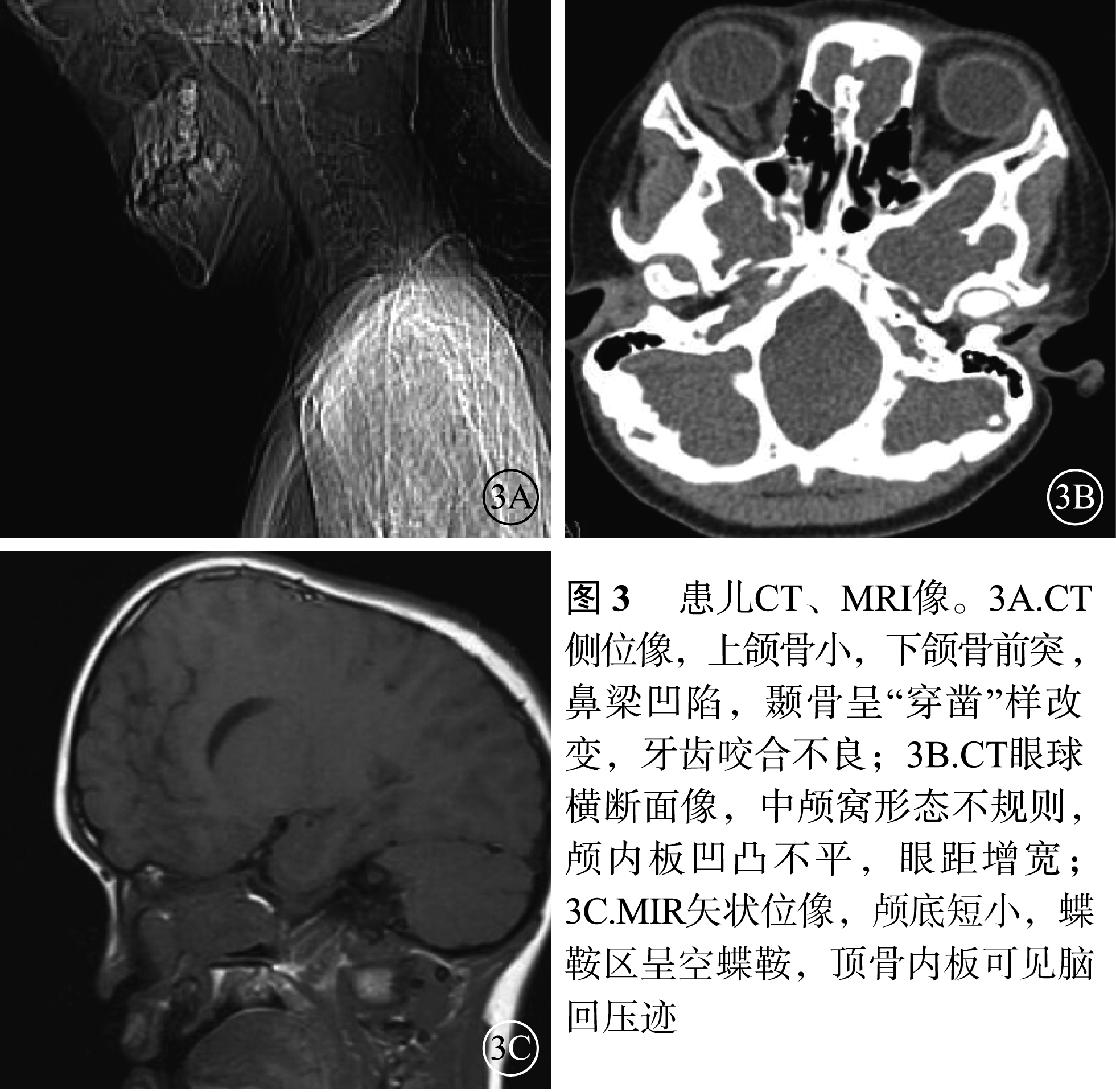
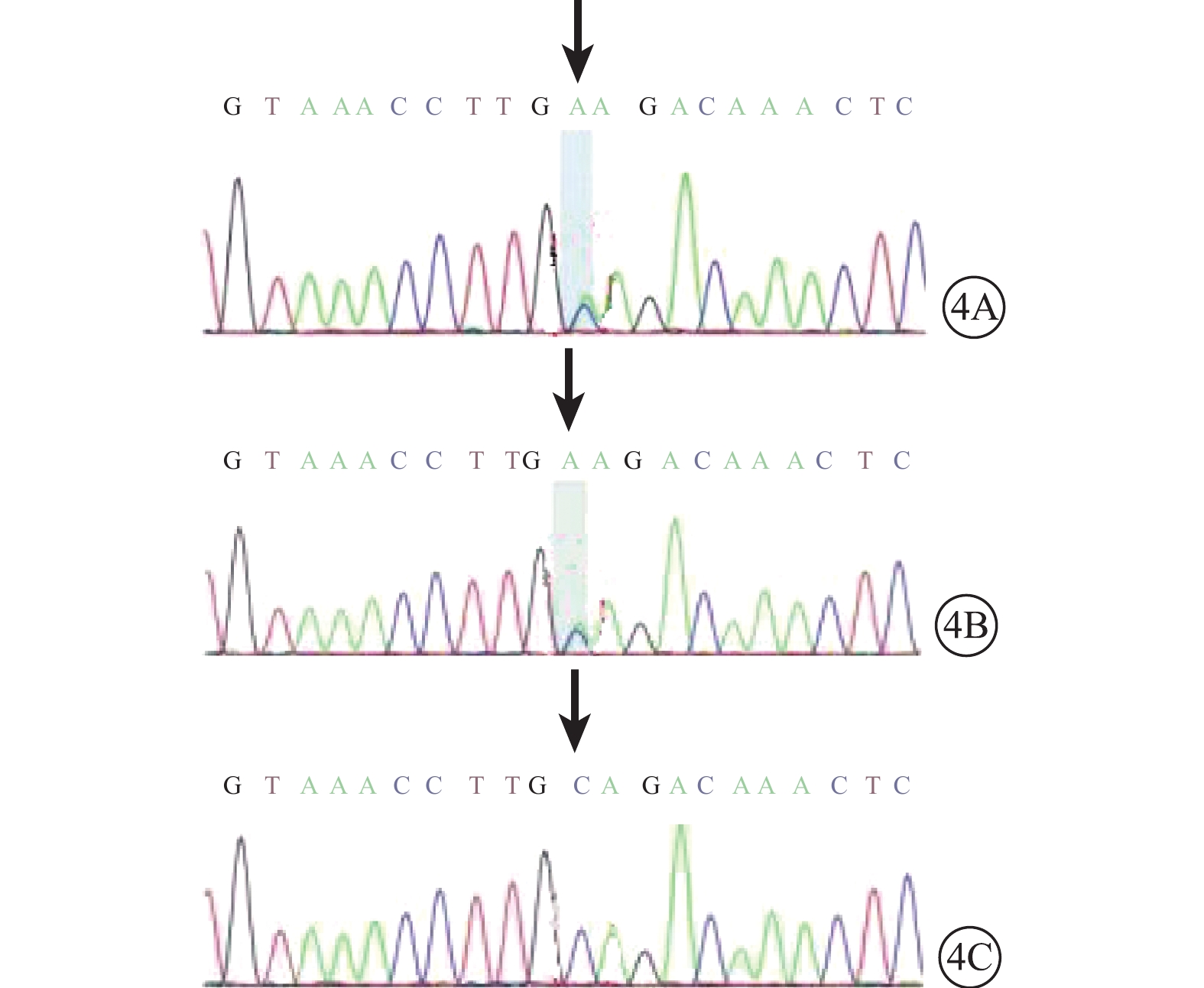 圖4
基因測序圖。4A. 患兒;4B. 父親;4C. 母親。患兒和其父親FGFR2基因chr10:123279599 p.Cys278Phe存在c.833G>T的雜合突變(黑箭);其母親改位點未見突變(黑箭)
圖4
基因測序圖。4A. 患兒;4B. 父親;4C. 母親。患兒和其父親FGFR2基因chr10:123279599 p.Cys278Phe存在c.833G>T的雜合突變(黑箭);其母親改位點未見突變(黑箭)
討論 Crouzon綜合征屬于顱縫早閉癥,以顱骨縫閉合過早、上頜發育不良及眼球突出等為主要特征[1]。臨床上根據其表現分為上頜型、顏面型、顱型、顱面型及假性Crouzon綜合征[2]。本例患兒顱面發育異常,但智力發育正常,根據基因檢測結果和臨床表現屬于上述5型中的顏面型。
本病可繼發壓迫性視神經病變,若未能及時治療,可出現視神經萎縮、視力下降[3]。本例患兒右眼視力下降2個月,就診時檢查未發現斜視,雙眼矯正視力右眼手光\?1.00×45→0.16,左眼?1.75\?1.50×90→0.9,可排除弱視。OCT檢查發現雙眼視盤顳側RNFL厚度變薄,與上述文獻報道相似。導致視神經病變的主要原因為腦水腫及顱內高壓[4],但本例患兒影像檢查并未發現腦水腫。其原因我們考慮可能與患兒顱面骨發育不對稱,導致右眼視神經壓迫較重進而視力影響較嚴重有關。
本病多為常染色體顯性遺傳,突變基因定位于染色體10q25-q26的FGFR2的基因片段,導致酪氨酸激酶及蛋白質編碼異常[5];少部分位于FGFR3基因片段[6, 7]。本例患兒和其父FGFR2基因存在c.833G>T chr10:123279599 p.Cys278Phe雜合突變;其母該位點未發現突變。該樣本線粒體基因相關熱點未發現異常,排除Leber遺傳性視神經病變等線粒體遺傳性疾病。
患兒男,12歲。因右眼視力下降2個月于2017年1月9日來我院眼科就診。患兒出生時即有雙眼突出,眼距較寬;智力發育正常。全身檢查:頭顱畸形、前額突出、鼻梁凹陷呈鸚鵡嘴狀鼻(圖1A);其他全身檢查未見異常。家族中其父親與患兒有相同特殊面容(圖1B);否認家族中有其他類似病例。眼部檢查:右眼視力0.08,手光/?1.00×45→0.16;左眼視力0.3,?1.75/?1.50×90→0.9。右眼眼壓15 mmHg(1 mmHg=0.133 kPa),左眼眼壓17 mmHg。雙眼眼位正;眼球突出度均為23 mm,眶距105 mm。輕度眼球震顫。雙眼眼前節檢查正常;瞳孔直接、間接對光反射正常。雙眼視盤邊界清楚,顏色略淡;視網膜血管走形、黃斑中心凹反光正常。光相干斷層掃描(OCT)檢查,雙眼視盤顳側神經纖維層(RNFL)厚度變薄(圖2A,2B)。雙眼視野、視覺誘發電位、色覺檢查均未見異常。耳聲阻抗檢查,左耳B形曲線,中耳積液;右耳正常。聽力檢查,左耳骨傳導下降;右耳正常。視神經孔、眼眶及頭部CT檢查,上頜骨小,下頜骨前突,鼻梁骨凹陷(圖3A);顱中窩形態不規則,眼距增寬,眼球略突出(圖3B);雙側視神經孔略增粗。頭部MRI檢查,左側放射冠區脫髓鞘;雙側篩竇及上頜竇炎;小腦扁桃體下疝畸形(圖3C)。其父親右眼視力1.0,左眼視力0.6。眼前節、眼底、OCT檢查結果均正常。基因檢查,患兒、其父親均發現耳聾相關基因成纖維細胞生長因子受體2(FGFR2)基因存在c.833G>T chr10:123279599 p.Cys278Phe雜合突變(圖4A,4B);其母親檢查結果正常(圖4C)。家系驗證結果顯示該突變來自患兒父親。線粒體基因組基因檢測,線粒體基因相關熱點突變未發現明確異常。診斷:Crouzon綜合征。
圖1
患兒和其父親面貌正面彩色像。1A. 患兒;1B. 父親。眼距增寬,眼球突出,鼻根低平
圖2
患兒雙眼視盤OCT像。2A. 右眼;2B. 左眼。顳側RNFL厚度變薄
圖4
基因測序圖。4A. 患兒;4B. 父親;4C. 母親。患兒和其父親FGFR2基因chr10:123279599 p.Cys278Phe存在c.833G>T的雜合突變(黑箭);其母親改位點未見突變(黑箭)
討論 Crouzon綜合征屬于顱縫早閉癥,以顱骨縫閉合過早、上頜發育不良及眼球突出等為主要特征[1]。臨床上根據其表現分為上頜型、顏面型、顱型、顱面型及假性Crouzon綜合征[2]。本例患兒顱面發育異常,但智力發育正常,根據基因檢測結果和臨床表現屬于上述5型中的顏面型。
本病可繼發壓迫性視神經病變,若未能及時治療,可出現視神經萎縮、視力下降[3]。本例患兒右眼視力下降2個月,就診時檢查未發現斜視,雙眼矯正視力右眼手光\?1.00×45→0.16,左眼?1.75\?1.50×90→0.9,可排除弱視。OCT檢查發現雙眼視盤顳側RNFL厚度變薄,與上述文獻報道相似。導致視神經病變的主要原因為腦水腫及顱內高壓[4],但本例患兒影像檢查并未發現腦水腫。其原因我們考慮可能與患兒顱面骨發育不對稱,導致右眼視神經壓迫較重進而視力影響較嚴重有關。
本病多為常染色體顯性遺傳,突變基因定位于染色體10q25-q26的FGFR2的基因片段,導致酪氨酸激酶及蛋白質編碼異常[5];少部分位于FGFR3基因片段[6, 7]。本例患兒和其父FGFR2基因存在c.833G>T chr10:123279599 p.Cys278Phe雜合突變;其母該位點未發現突變。該樣本線粒體基因相關熱點未發現異常,排除Leber遺傳性視神經病變等線粒體遺傳性疾病。