引用本文: 付燕, 顧朝輝, 石笑楠. Waardenburg綜合征一例眼部表現. 中華眼底病雜志, 2018, 34(1): 75-76. doi: 10.3760/cma.j.issn.1005-1015.2018.01.021 復制
患兒女,9歲。因雙眼視物不清1年余,2017年2月28日來我院眼科就診。足月,剖腹產,出生體重3100 g。無吸氧史。自幼發現雙眼虹膜異色。出生3個月時,患兒家長發現患兒雙耳失聰,于2011年在外院診斷為雙側極重度感音神經性耳聾并行右耳經面隱窩入路人工耳蝸植入手術。語言及運動發育均滯后。母親從事油漆涂料有關工作,懷孕期間有發熱服藥史,具體不詳。父母身體健康,非近親婚配。其父系及母系親屬無同樣疾患者。2013年,患兒及其父母經線粒體12srRNA 1494位點、1555位點及SLC基因IVS7-2、GJB2基因235位點檢測,未發現突變。
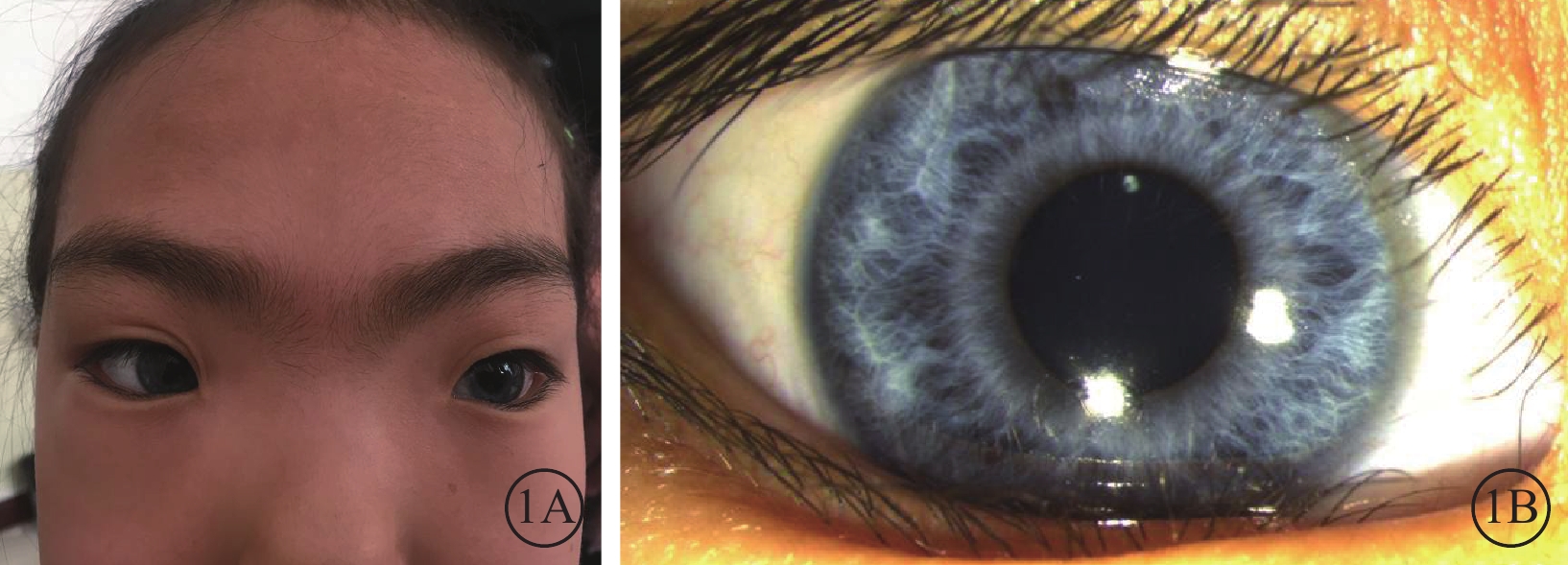
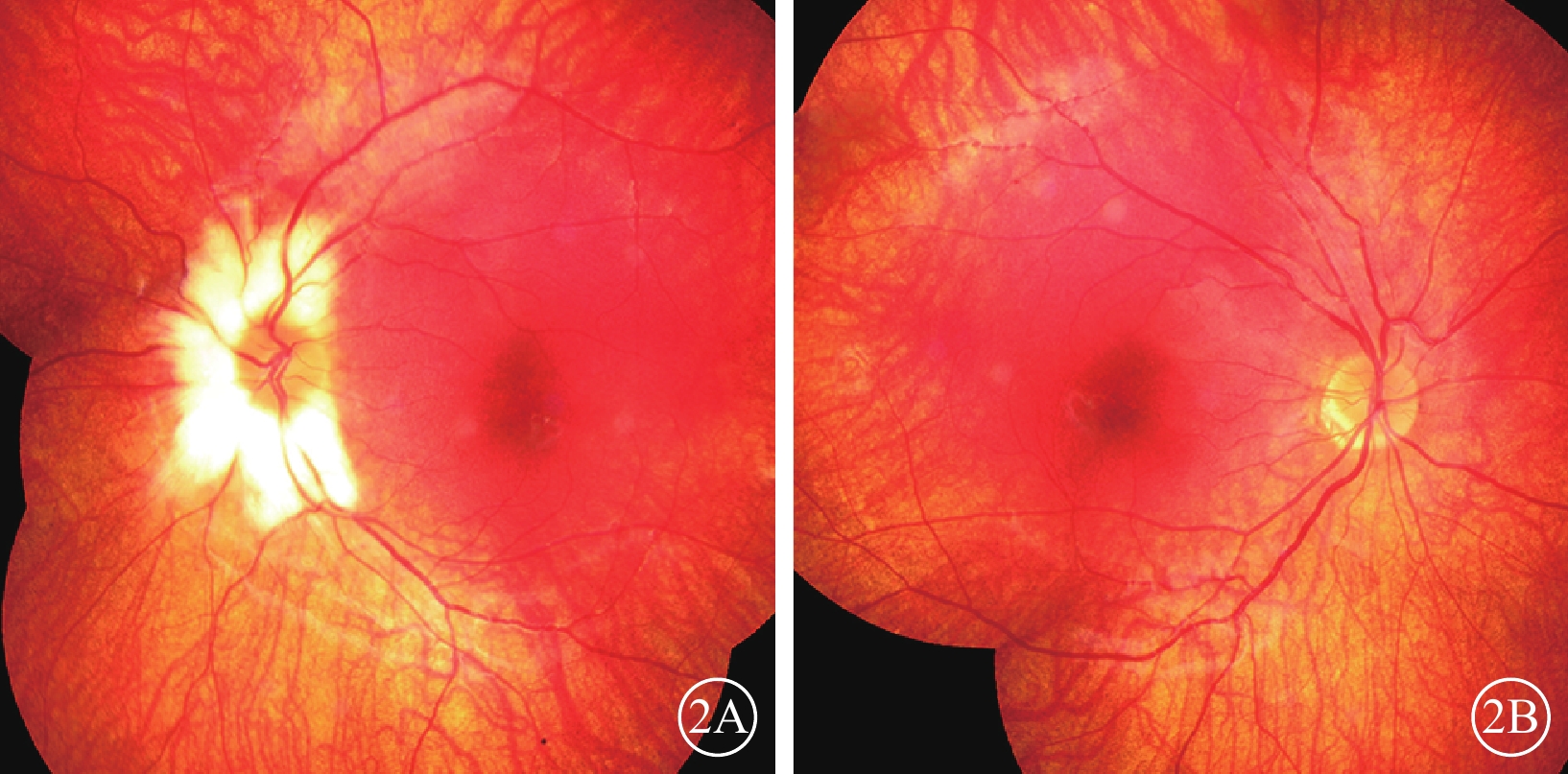
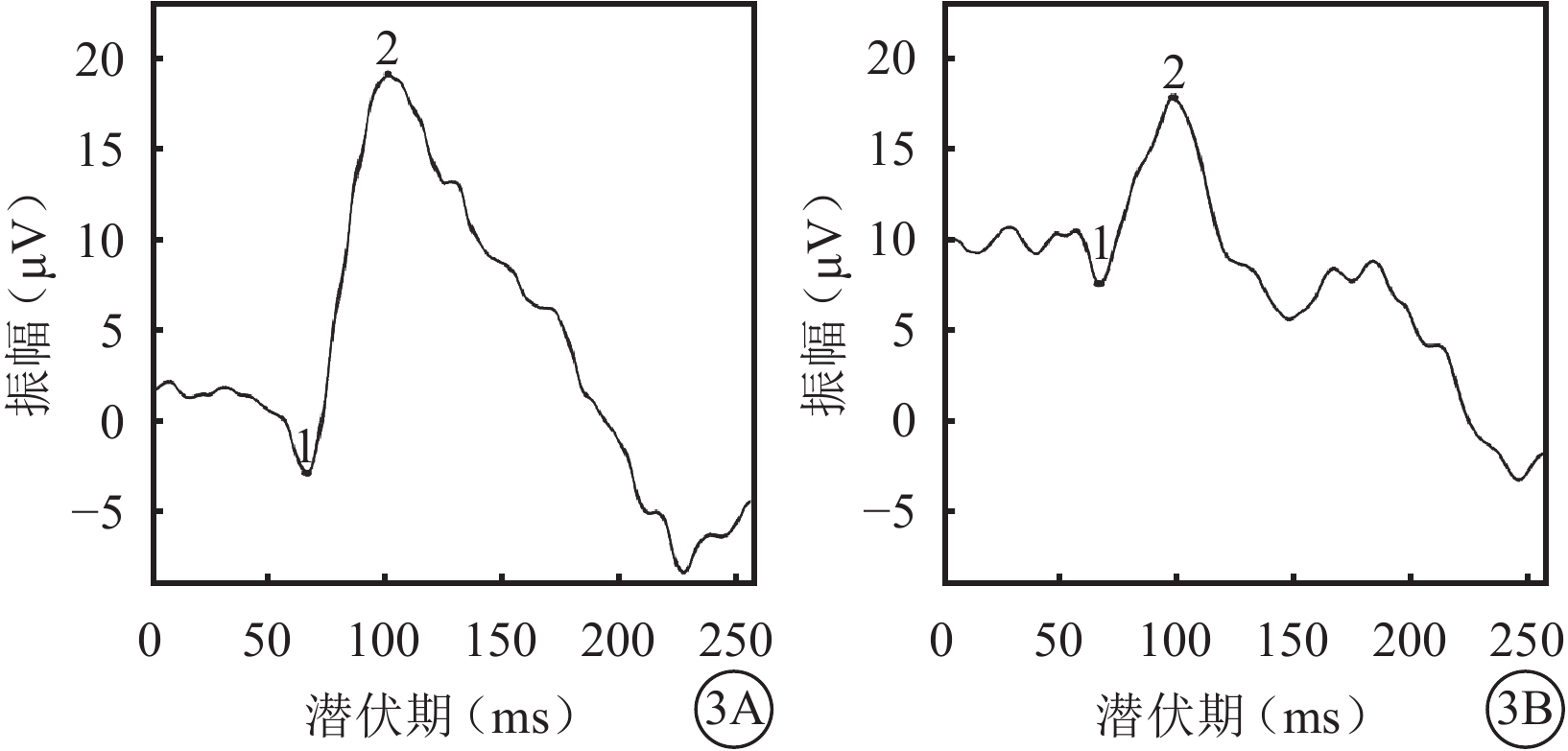
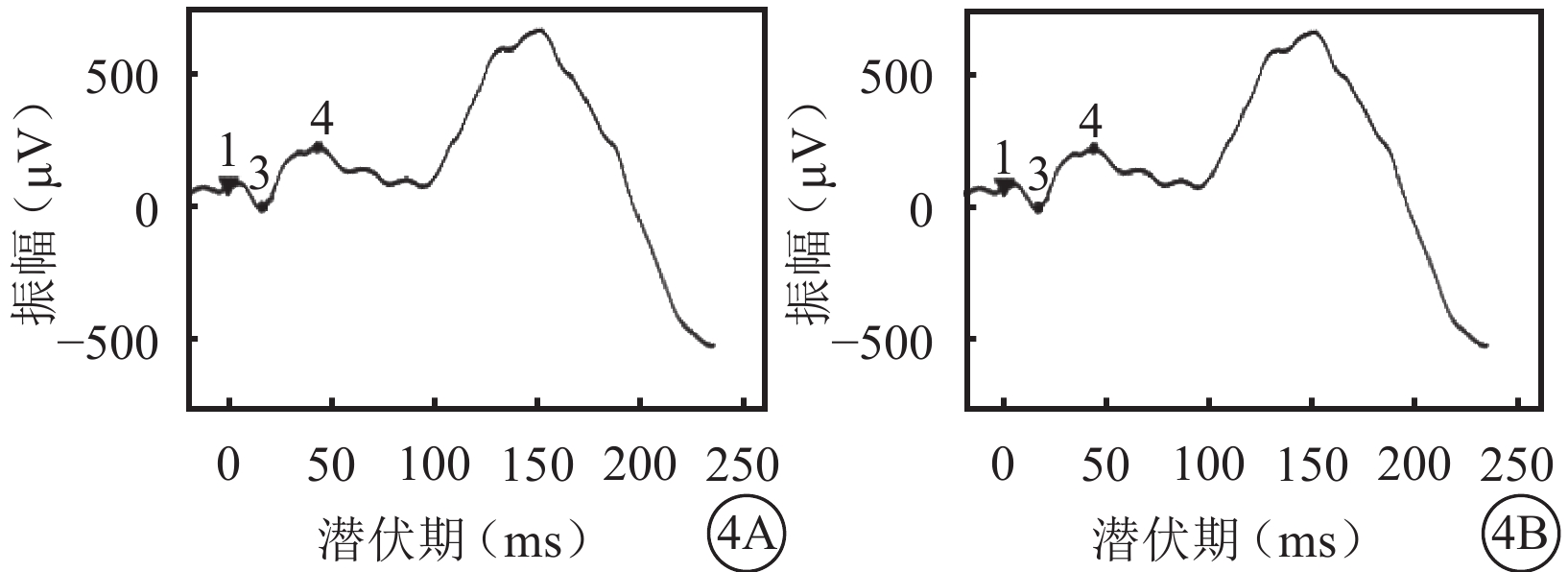
體格檢查:全身皮膚及毛發色澤無異常。面容呈特殊方形,面部鼻根粗大,鼻額角消失,內側眉毛致密(圖1A)。眼部檢查:右眼視力0.12,矯正視力?7.25 DS→0.5;左眼視力0.2,矯正視力?6.5 DS→0.5。色覺正常。內眥間距45 mm,外眥間距87 mm,雙瞼裂長21 mm。淚小點外移,淚道沖洗通暢。眼位交替遮蓋,外→中?20°。雙眼角膜透明,前房清晰,中深,瞳孔圓,對光反射靈敏;雙側虹膜色素脫失呈灰白色,虹膜基質萎縮(圖1B),晶狀體及玻璃體未見明顯混濁。眼底檢查,右眼視盤周圍羽毛狀有髓鞘神經纖維,范圍約1個視盤直徑;左眼視盤正常。雙眼眼底橙紅色,視網膜色素上皮(RPE)萎縮伴色素脫失,暴露脈絡膜大中血管,視網膜血管走形無明顯異常,黃斑中心凹反光不清(圖2)。雙眼視盤及黃斑光相干斷層掃描(OCT)檢查未見明顯異常。視覺誘發電位(VEP)檢查,雙眼P100波潛伏期延長,振幅降低(圖3)。視網膜電圖(ERG)檢查,a、b波及振蕩電位(OPs)峰時延遲,振幅降低(圖4)。診斷:(1)Waardenburg綜合征(WS);(2)雙眼屈光不正;(3)雙眼弱視;(4)雙眼外斜視。給予患兒配鏡矯正屈光不正,密切隨訪。建議患兒完善基因檢查。
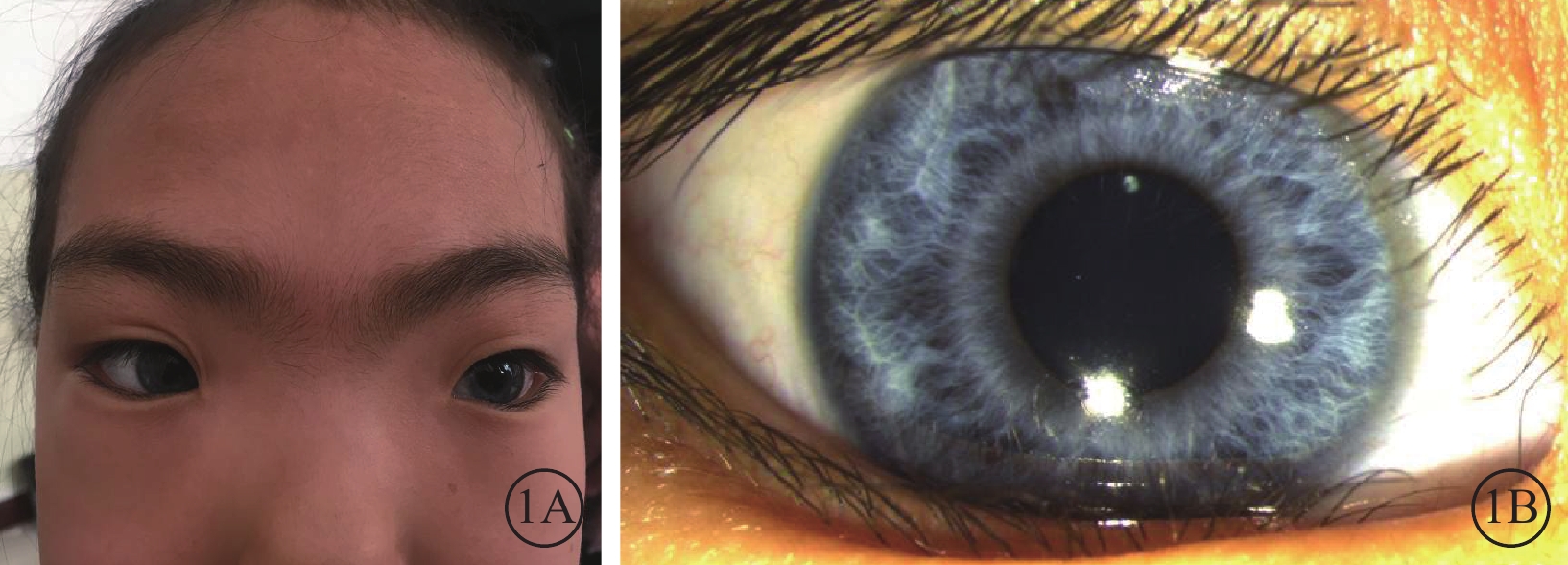 圖1
患兒面部外觀像及右眼眼前節像。1A. 面部外觀像,面容呈特殊方形,面部鼻根粗大,鼻額角消失,內側眉毛致密;1B. 右眼眼前節像,虹膜色素脫失呈灰白色,虹膜基質萎縮
圖1
患兒面部外觀像及右眼眼前節像。1A. 面部外觀像,面容呈特殊方形,面部鼻根粗大,鼻額角消失,內側眉毛致密;1B. 右眼眼前節像,虹膜色素脫失呈灰白色,虹膜基質萎縮
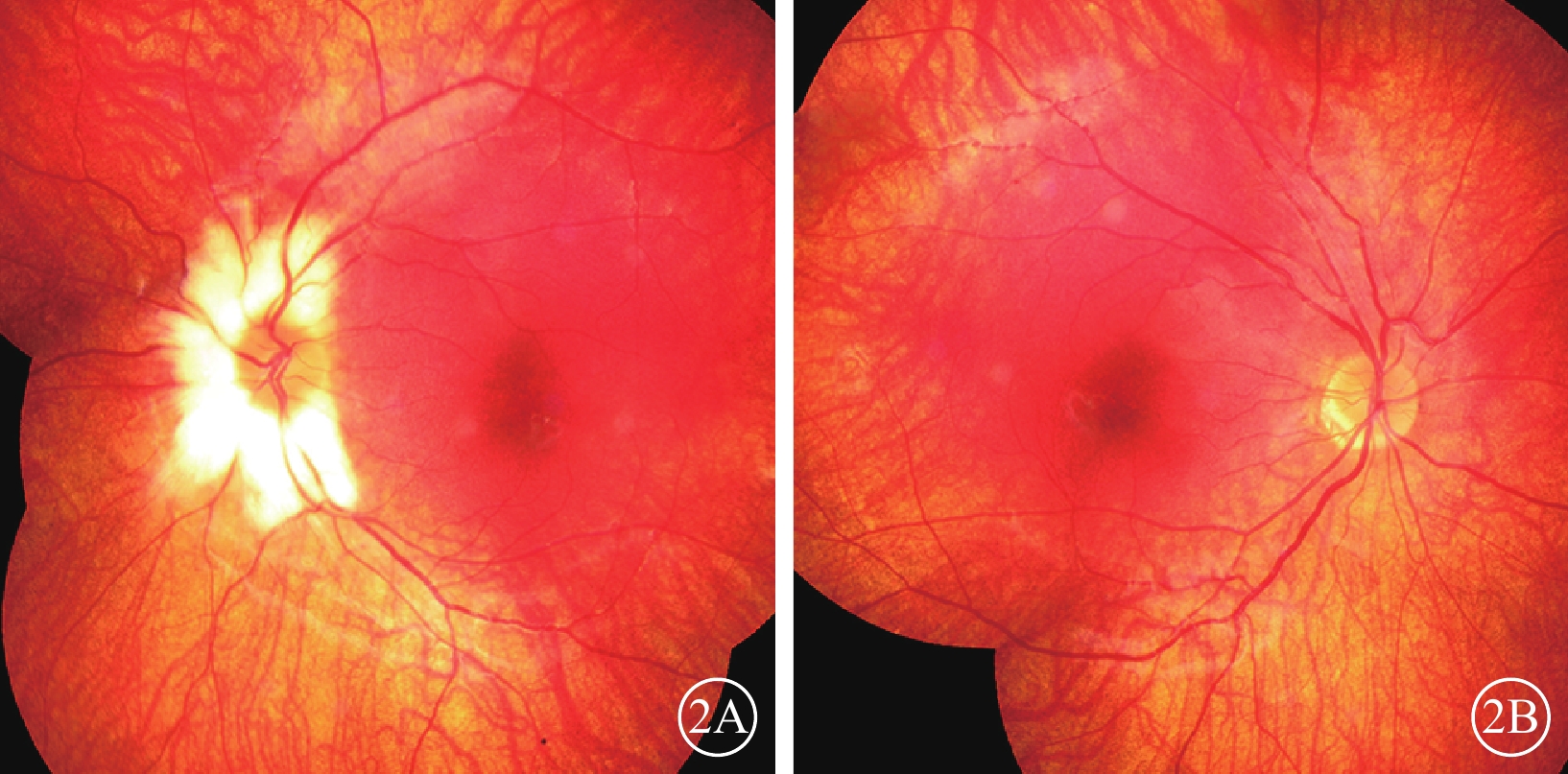 圖2
雙眼彩色眼底像。2A. 左眼;2B. 右眼。眼底呈橙紅色,視網膜色素減少,視網膜血管走形正常
圖2
雙眼彩色眼底像。2A. 左眼;2B. 右眼。眼底呈橙紅色,視網膜色素減少,視網膜血管走形正常
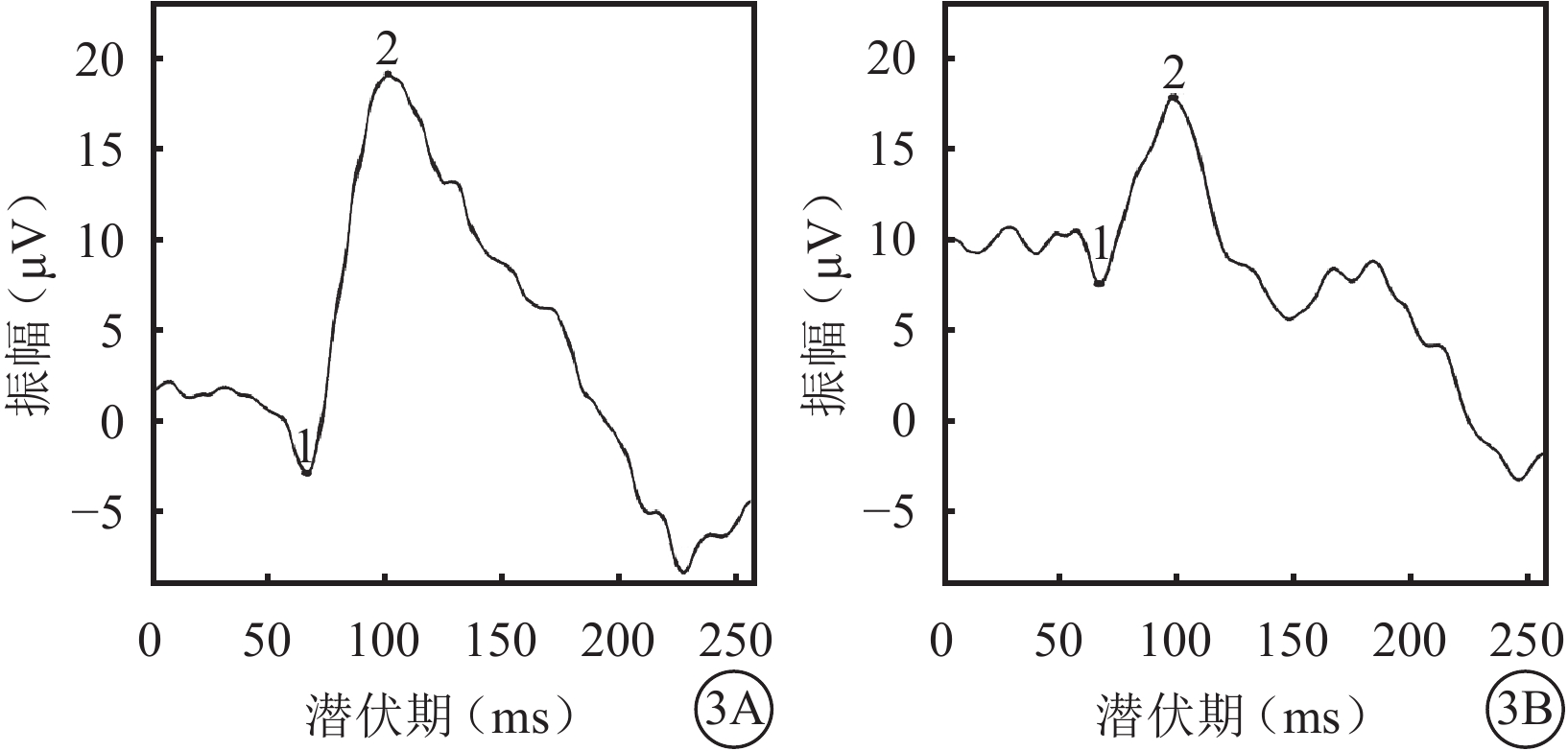 圖3
雙眼VEP像。3A. 右眼;3B. 左眼。雙眼P100波潛伏期延長,振幅降低
圖3
雙眼VEP像。3A. 右眼;3B. 左眼。雙眼P100波潛伏期延長,振幅降低
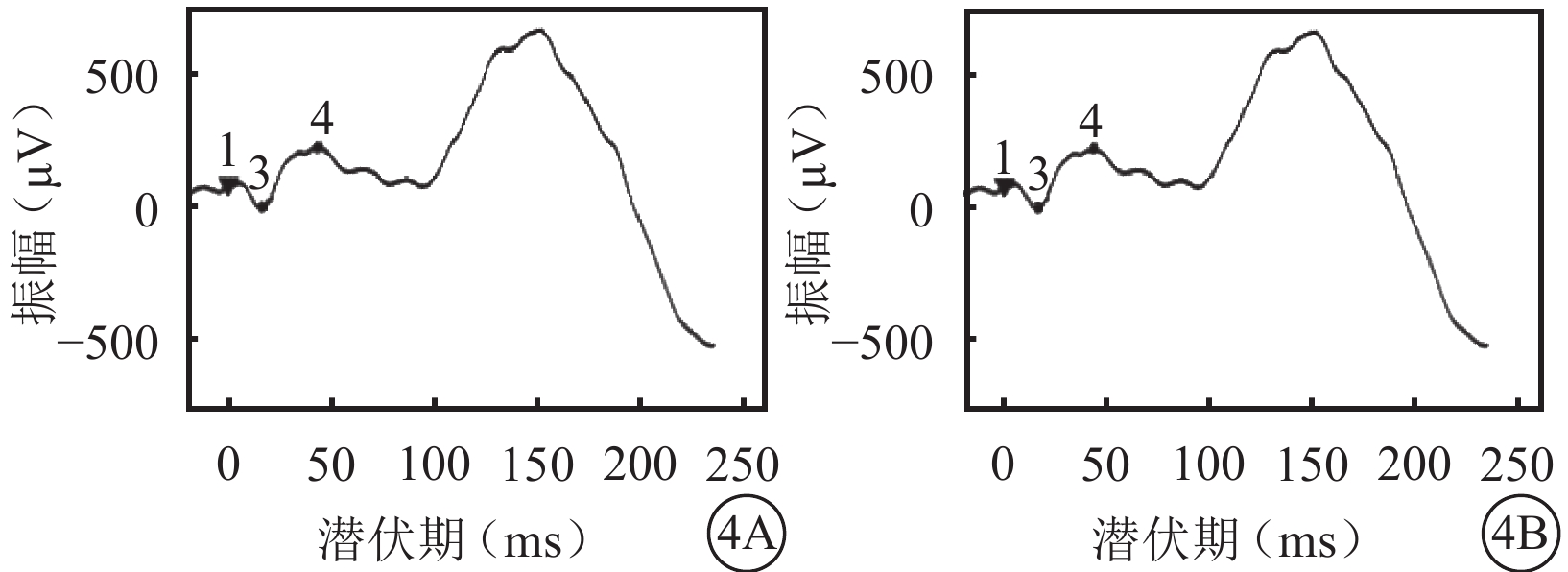 圖4
雙眼ERG像。4A. 右眼;4B. 左眼。a、b波及OPs峰時延遲,振幅降低
圖4
雙眼ERG像。4A. 右眼;4B. 左眼。a、b波及OPs峰時延遲,振幅降低
討論 WS是罕見的以先天性感音神經性耳聾及皮膚、虹膜、毛發的色素分布異常為主要特征的遺傳綜合征[1]。根據患兒不同的臨床表現,可將WS分為WS1、WS2、WS3及WS4型[2]。WS1型先天性感音神經性耳聾、虹膜色素分布異常、內眥異位。次要標準:先天性白斑病、<30歲白發、連眉或眉心毛發旺盛、鼻根高或寬、鼻翼發育不全;WS2型先天性感音神經性耳聾、色素異常,無內眥異位;WS3型:WS1型癥狀合并肌肉骨路發育異常;WS4型:WS2型癥狀合并先天性巨結腸或胃腸道閉鎖。在WS的主要臨床表型中,約20%~55%伴有重度或極重度感音神經性耳聾。
WS眼部最具特征性的表現是內眥外移,常伴隨著瞼裂縮小、淚小點外移、內眥贅皮、眼距過寬、小角膜。其次是部分或全部虹膜呈灰色、藍白色或亮藍色,眼底色素異常、脫失或色素斑點,視網膜形成不全,視神經發育不全等。斜視在WS1型患者中的發生率較正常人群高[3]。本例患兒雙側極重度感音神經性耳聾,內側眉毛粗重幾乎連眉,鼻根部粗大,內眥外移,淚小點外移,虹膜灰白色,眼底視網膜色素減少。提示患兒可能為WS1型。目前對WS的治療主要是對癥治療。本例患兒雙眼弱視,給予戴鏡矯正屈光不正。
WS具有遺傳異質性,不同基因的突變可導致不同類型的WS,目前發現與WS發病有關的基因共6個,包括轉錄因子PAX3、MITF、SNAI2、SOX10和信號分子ENDR3和EDN3。其中PAX3是WS1、WS3型的主要致病基因[4]。WS2型的遺傳基礎較復雜,MITF、SNAI2、SOX10和ENDR3基因突變均與WS2相關。ENDR3和EDN3和SOX10基因突變主要與WS4有關。本例患者很可能屬WS1型,建議患兒及其父母完善基因檢測,便于產前咨詢及評定下一代的遺傳風險。但因患兒父親常年在外,暫未完成基因檢測,目前密切隨訪中。
患兒女,9歲。因雙眼視物不清1年余,2017年2月28日來我院眼科就診。足月,剖腹產,出生體重3100 g。無吸氧史。自幼發現雙眼虹膜異色。出生3個月時,患兒家長發現患兒雙耳失聰,于2011年在外院診斷為雙側極重度感音神經性耳聾并行右耳經面隱窩入路人工耳蝸植入手術。語言及運動發育均滯后。母親從事油漆涂料有關工作,懷孕期間有發熱服藥史,具體不詳。父母身體健康,非近親婚配。其父系及母系親屬無同樣疾患者。2013年,患兒及其父母經線粒體12srRNA 1494位點、1555位點及SLC基因IVS7-2、GJB2基因235位點檢測,未發現突變。
體格檢查:全身皮膚及毛發色澤無異常。面容呈特殊方形,面部鼻根粗大,鼻額角消失,內側眉毛致密(圖1A)。眼部檢查:右眼視力0.12,矯正視力?7.25 DS→0.5;左眼視力0.2,矯正視力?6.5 DS→0.5。色覺正常。內眥間距45 mm,外眥間距87 mm,雙瞼裂長21 mm。淚小點外移,淚道沖洗通暢。眼位交替遮蓋,外→中?20°。雙眼角膜透明,前房清晰,中深,瞳孔圓,對光反射靈敏;雙側虹膜色素脫失呈灰白色,虹膜基質萎縮(圖1B),晶狀體及玻璃體未見明顯混濁。眼底檢查,右眼視盤周圍羽毛狀有髓鞘神經纖維,范圍約1個視盤直徑;左眼視盤正常。雙眼眼底橙紅色,視網膜色素上皮(RPE)萎縮伴色素脫失,暴露脈絡膜大中血管,視網膜血管走形無明顯異常,黃斑中心凹反光不清(圖2)。雙眼視盤及黃斑光相干斷層掃描(OCT)檢查未見明顯異常。視覺誘發電位(VEP)檢查,雙眼P100波潛伏期延長,振幅降低(圖3)。視網膜電圖(ERG)檢查,a、b波及振蕩電位(OPs)峰時延遲,振幅降低(圖4)。診斷:(1)Waardenburg綜合征(WS);(2)雙眼屈光不正;(3)雙眼弱視;(4)雙眼外斜視。給予患兒配鏡矯正屈光不正,密切隨訪。建議患兒完善基因檢查。
圖1
患兒面部外觀像及右眼眼前節像。1A. 面部外觀像,面容呈特殊方形,面部鼻根粗大,鼻額角消失,內側眉毛致密;1B. 右眼眼前節像,虹膜色素脫失呈灰白色,虹膜基質萎縮
圖2
雙眼彩色眼底像。2A. 左眼;2B. 右眼。眼底呈橙紅色,視網膜色素減少,視網膜血管走形正常
圖3
雙眼VEP像。3A. 右眼;3B. 左眼。雙眼P100波潛伏期延長,振幅降低
圖4
雙眼ERG像。4A. 右眼;4B. 左眼。a、b波及OPs峰時延遲,振幅降低
討論 WS是罕見的以先天性感音神經性耳聾及皮膚、虹膜、毛發的色素分布異常為主要特征的遺傳綜合征[1]。根據患兒不同的臨床表現,可將WS分為WS1、WS2、WS3及WS4型[2]。WS1型先天性感音神經性耳聾、虹膜色素分布異常、內眥異位。次要標準:先天性白斑病、<30歲白發、連眉或眉心毛發旺盛、鼻根高或寬、鼻翼發育不全;WS2型先天性感音神經性耳聾、色素異常,無內眥異位;WS3型:WS1型癥狀合并肌肉骨路發育異常;WS4型:WS2型癥狀合并先天性巨結腸或胃腸道閉鎖。在WS的主要臨床表型中,約20%~55%伴有重度或極重度感音神經性耳聾。
WS眼部最具特征性的表現是內眥外移,常伴隨著瞼裂縮小、淚小點外移、內眥贅皮、眼距過寬、小角膜。其次是部分或全部虹膜呈灰色、藍白色或亮藍色,眼底色素異常、脫失或色素斑點,視網膜形成不全,視神經發育不全等。斜視在WS1型患者中的發生率較正常人群高[3]。本例患兒雙側極重度感音神經性耳聾,內側眉毛粗重幾乎連眉,鼻根部粗大,內眥外移,淚小點外移,虹膜灰白色,眼底視網膜色素減少。提示患兒可能為WS1型。目前對WS的治療主要是對癥治療。本例患兒雙眼弱視,給予戴鏡矯正屈光不正。
WS具有遺傳異質性,不同基因的突變可導致不同類型的WS,目前發現與WS發病有關的基因共6個,包括轉錄因子PAX3、MITF、SNAI2、SOX10和信號分子ENDR3和EDN3。其中PAX3是WS1、WS3型的主要致病基因[4]。WS2型的遺傳基礎較復雜,MITF、SNAI2、SOX10和ENDR3基因突變均與WS2相關。ENDR3和EDN3和SOX10基因突變主要與WS4有關。本例患者很可能屬WS1型,建議患兒及其父母完善基因檢測,便于產前咨詢及評定下一代的遺傳風險。但因患兒父親常年在外,暫未完成基因檢測,目前密切隨訪中。