引用本文: 趙娟, 閆博婧, 吳志中, 李根林. 先天性黃斑缺損兄弟二例. 中華眼底病雜志, 2017, 33(6): 646-647. doi: 10.3760/cma.j.issn.1005-1015.2017.06.025 復制
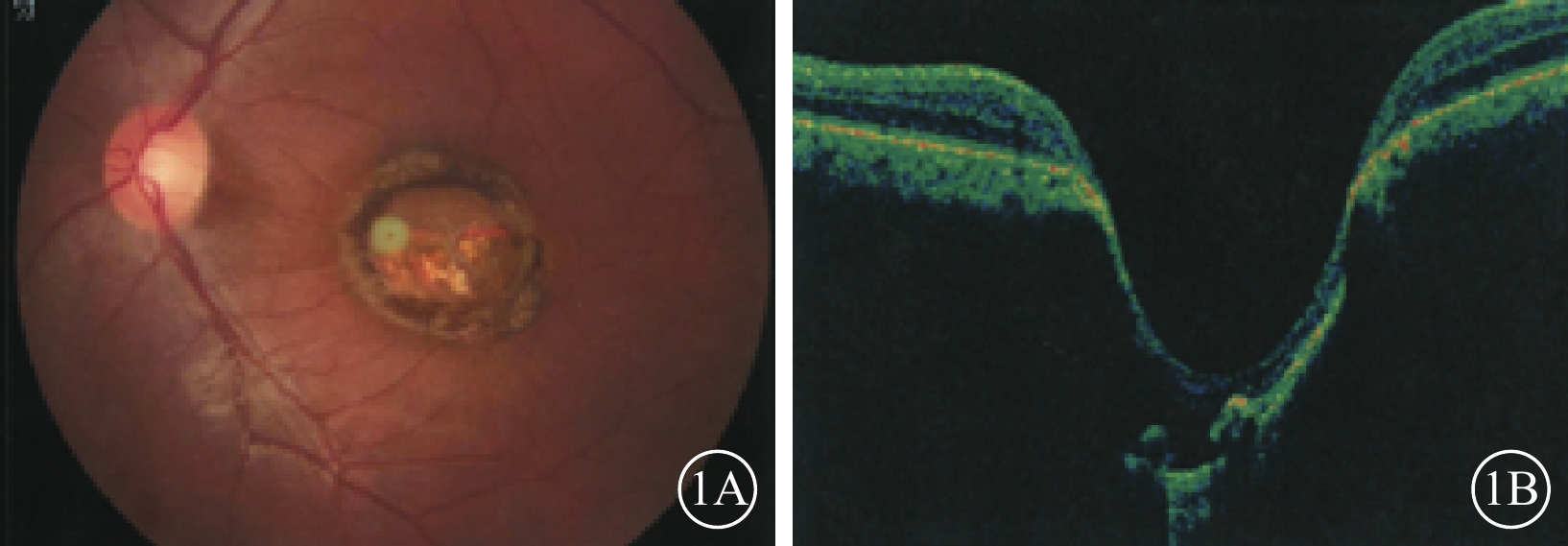
例1 患者男,24歲。因自幼雙眼視力差于2016年12月來我院眼科就診。父母非近親結婚,除其兄自幼雙眼視力差外,余家族史無特殊。否認夜盲及全身病史、母親孕期感染及藥物使用史。眼科檢查:雙眼眼位正,無眼球震顫。雙眼視力0.1,均不能矯正。右眼眼壓18 mmHg(1 mmHg=0.133 kPa),左眼眼壓19 mmHg。雙眼瞳孔對光反射遲鈍,其余眼前節檢查正常。右眼視盤邊界清楚,顏色基本正常,杯盤比(C/D)=0.3;視網膜平復,黃斑區可見約1/5個視盤直徑(DD)大小的橫橢圓形缺損區,其邊界清晰,中間透見脈絡膜血管。左眼視盤邊界清楚,顏色淡黃,C/D=0.3;視網膜平復,黃斑區可見約1.5 DD大小的類圓形缺損區(圖1A);其邊界清晰,凹陷深,中間可見灰白色鞏膜組織,透見脈絡膜大血管,病變邊緣可見色素沉著。光相干斷層掃描(OCT)檢查,右眼黃斑缺損區視網膜外層結構缺失,橢圓體帶缺損,視網膜色素上皮(RPE)層完整;左眼黃斑缺損區視網膜脈絡膜組織隨鞏膜組織局限性向外凹陷,凹陷區域內視網膜神經上皮層明顯變薄,部分缺損,其下方可見淺脫離(圖1B)。基因檢查:患者攜帶下列雜合突變:(1)RP9 c. 448C>T,p. R150*;(2)VSX1 c. 479G>T,p. G160V;(3)CEP290 c. 3573+3A>G;(4)CEP290 c. 366A>G,p. K122K。診斷:雙眼先天性黃斑缺損(色素型)。
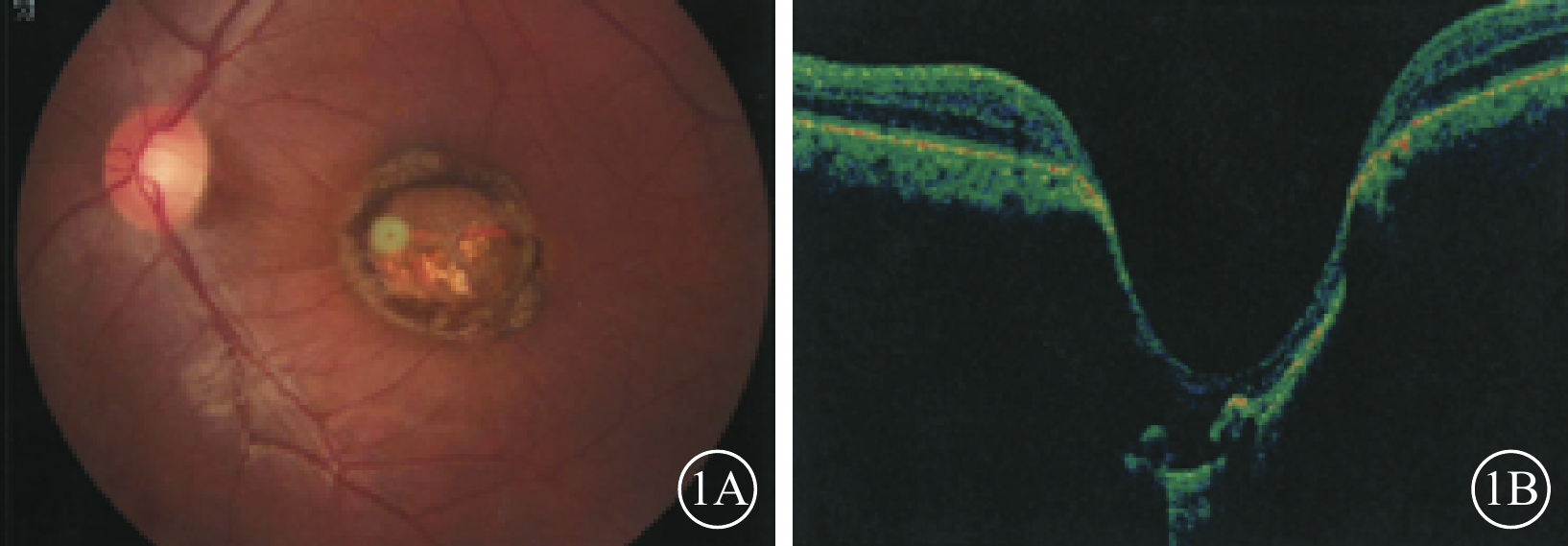 圖1
例1患者左眼彩色眼底、OCT像。1A. 彩色眼底像,黃斑區可見一大小約1.5 DD的類圓形缺損區,邊界清晰,病灶邊緣存在色素沉著;1B. OCT像,黃斑缺損區視網膜脈絡膜組織局限性向外凹陷,凹陷區域內視網膜神經上皮層可見部分缺損
圖1
例1患者左眼彩色眼底、OCT像。1A. 彩色眼底像,黃斑區可見一大小約1.5 DD的類圓形缺損區,邊界清晰,病灶邊緣存在色素沉著;1B. OCT像,黃斑缺損區視網膜脈絡膜組織局限性向外凹陷,凹陷區域內視網膜神經上皮層可見部分缺損
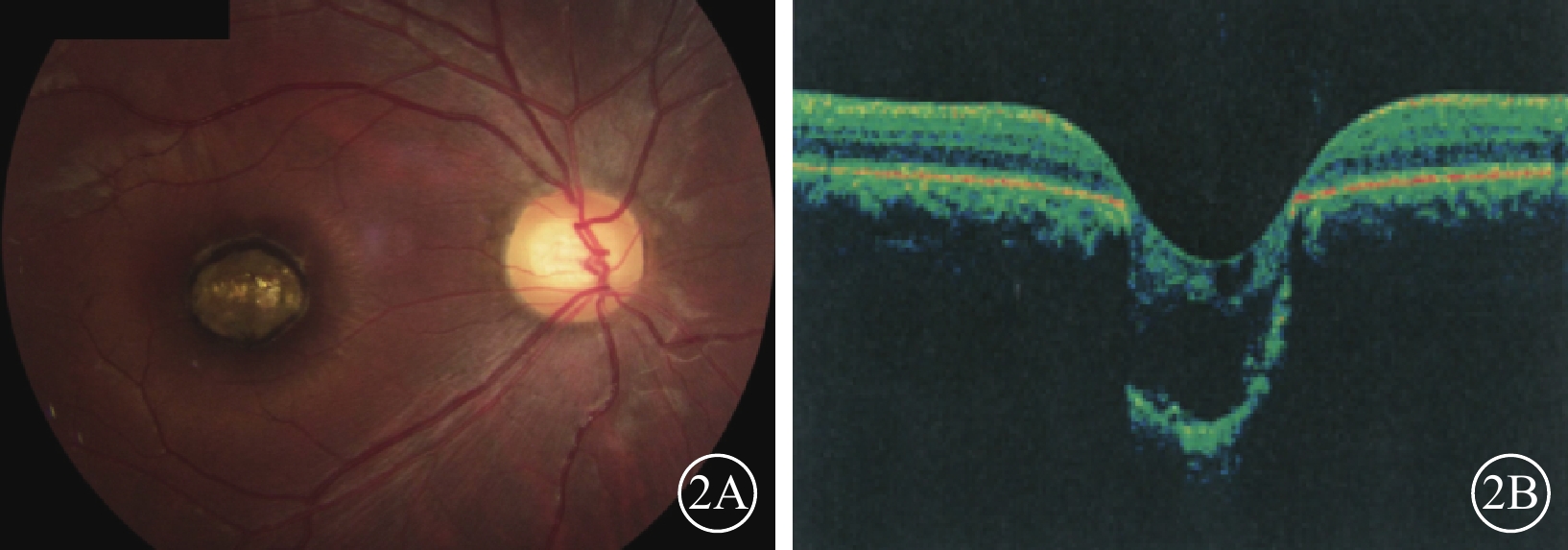 圖2
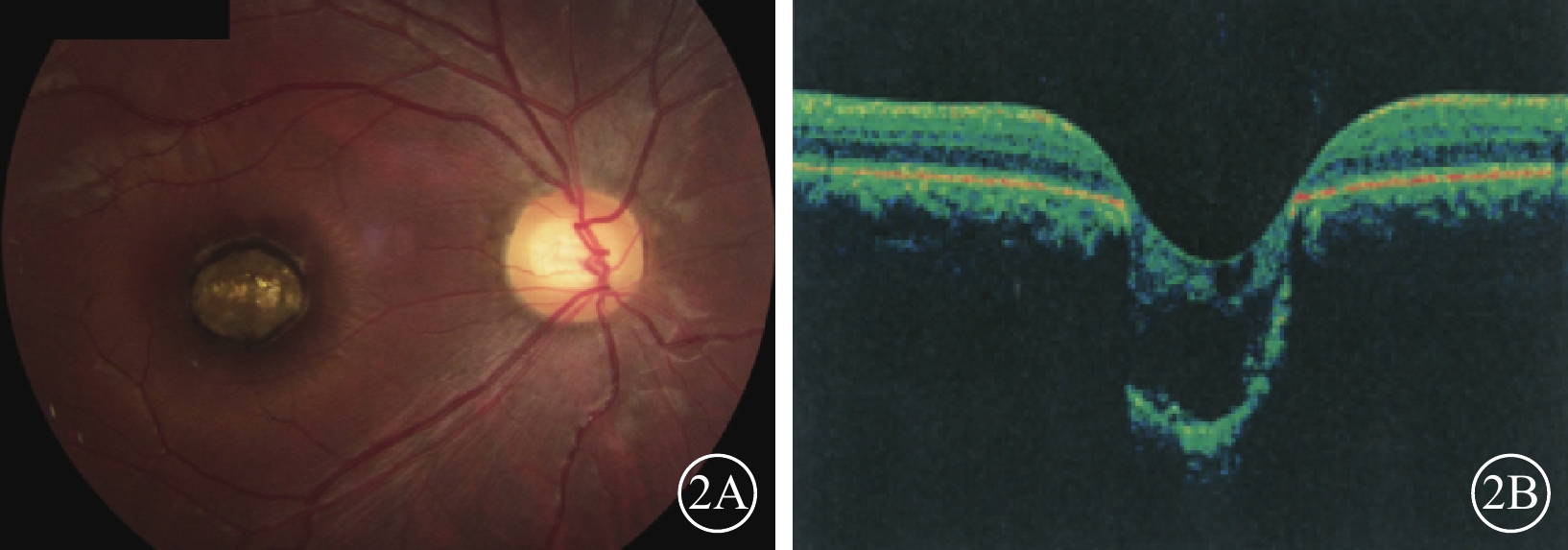
例2患者右眼彩色眼底、OCT像。2A. 彩色眼底像,黃斑區可見一大小約1.0 DD的邊界清晰的橢圓形缺損;2B. OCT像,病灶區視網膜脈絡膜組織隨鞏膜組織向外凹陷,凹陷區域內視網膜神經上皮層薄變及缺損,神經上皮層間呈囊樣變化伴淺脫離
圖2
例2患者右眼彩色眼底、OCT像。2A. 彩色眼底像,黃斑區可見一大小約1.0 DD的邊界清晰的橢圓形缺損;2B. OCT像,病灶區視網膜脈絡膜組織隨鞏膜組織向外凹陷,凹陷區域內視網膜神經上皮層薄變及缺損,神經上皮層間呈囊樣變化伴淺脫離
例2 例1之兄,29歲。因自幼雙眼視力差與例1同時來我院就診。眼科檢查,雙眼眼位正位,無眼球震顫。右眼視力0.1,?7.50 DS/+4.00 DC×90°=0.1;左眼視力0.05,?4.00 DS/+3.00 DC×85°=0.1。右眼眼壓19 mmHg,左眼眼壓20 mmHg。雙眼瞳孔對光反射遲鈍,其余眼前節檢查正常。雙眼視盤邊界清楚,顏色基本正常,C/D=0.3;視網膜平復,黃斑區可見約1.0 DD大小的類圓形缺損區(圖2A);其邊界清晰,凹陷深,中間可見灰白色鞏膜組織,脈絡膜毛細血管缺失。OCT檢查,雙眼黃斑缺損區視網膜脈絡膜組織隨鞏膜組織局限性向外凹陷,凹陷區域內視網膜神經上皮層變薄,部分缺損,神經上皮層間囊樣改變,其下見淺脫離(圖2B)。基因檢查:患者攜帶下列雜合突變:(1)CEP290 c. 3573+3A>G;(2)CEP290 c. 366A>G,p. K122K。診斷:雙眼先天性黃斑缺損(色素型)。
討論 先天性黃斑缺損多為常染色體顯性遺傳,少數為隱性遺傳,亦存在散發病例[1]。臨床表現為患眼中心視力差,注視不佳或斜視,眼底異常改變;病灶可位于黃斑或黃斑附近,表面平坦或輕中度凹陷,其大小、形狀和顏色呈不規則變化[2]。OCT檢查可反映黃斑缺損區組織結構的變化[3]。依據鞏膜暴露的程度及色素的多少本病可分為色素型、無色素型以及合并血管異常型[4]。本文報道的2例患者均可見黃斑區結構缺失伴色素沉著。根據OCT檢查并結合基因檢測結果,符合先天性黃斑缺損(色素型)診斷。2例患者的父母無臨床表現,但未行基因檢測,其發病的遺傳學特征有待進一步研究。
本病需與以下疾病鑒別:(1)Stargardt病:主要特征為黃斑區出現圓形或橢圓形的邊界清晰的黃色病灶,呈“牛眼”樣表現,病灶周圍及后極部可見黃色斑點[5];(2)脈絡膜缺損:脈絡膜局限性缺損常伴有其它眼部異常改變,如虹膜缺損、視神經發育不良或缺損、黃斑缺損等[6];(3)彌漫性脈絡膜營養障礙:后極部出現脈絡膜萎縮,繼而向視網膜周邊部緩慢進展,最終脈絡膜呈彌漫性萎縮,鞏膜暴露,無黃斑結構缺損;(4)中心性暈輪狀視網膜脈絡膜萎縮[7]:雙眼呈對稱性、邊界清晰的脈絡膜視網膜萎縮,萎縮區一般小于1.0 DD,無黃斑結構缺損。本文報道的2例患者眼底無“牛眼”樣改變及黃色斑點以及無虹膜、視神經缺損,可資鑒別。
例1 患者男,24歲。因自幼雙眼視力差于2016年12月來我院眼科就診。父母非近親結婚,除其兄自幼雙眼視力差外,余家族史無特殊。否認夜盲及全身病史、母親孕期感染及藥物使用史。眼科檢查:雙眼眼位正,無眼球震顫。雙眼視力0.1,均不能矯正。右眼眼壓18 mmHg(1 mmHg=0.133 kPa),左眼眼壓19 mmHg。雙眼瞳孔對光反射遲鈍,其余眼前節檢查正常。右眼視盤邊界清楚,顏色基本正常,杯盤比(C/D)=0.3;視網膜平復,黃斑區可見約1/5個視盤直徑(DD)大小的橫橢圓形缺損區,其邊界清晰,中間透見脈絡膜血管。左眼視盤邊界清楚,顏色淡黃,C/D=0.3;視網膜平復,黃斑區可見約1.5 DD大小的類圓形缺損區(圖1A);其邊界清晰,凹陷深,中間可見灰白色鞏膜組織,透見脈絡膜大血管,病變邊緣可見色素沉著。光相干斷層掃描(OCT)檢查,右眼黃斑缺損區視網膜外層結構缺失,橢圓體帶缺損,視網膜色素上皮(RPE)層完整;左眼黃斑缺損區視網膜脈絡膜組織隨鞏膜組織局限性向外凹陷,凹陷區域內視網膜神經上皮層明顯變薄,部分缺損,其下方可見淺脫離(圖1B)。基因檢查:患者攜帶下列雜合突變:(1)RP9 c. 448C>T,p. R150*;(2)VSX1 c. 479G>T,p. G160V;(3)CEP290 c. 3573+3A>G;(4)CEP290 c. 366A>G,p. K122K。診斷:雙眼先天性黃斑缺損(色素型)。
圖1
例1患者左眼彩色眼底、OCT像。1A. 彩色眼底像,黃斑區可見一大小約1.5 DD的類圓形缺損區,邊界清晰,病灶邊緣存在色素沉著;1B. OCT像,黃斑缺損區視網膜脈絡膜組織局限性向外凹陷,凹陷區域內視網膜神經上皮層可見部分缺損
圖2
例2患者右眼彩色眼底、OCT像。2A. 彩色眼底像,黃斑區可見一大小約1.0 DD的邊界清晰的橢圓形缺損;2B. OCT像,病灶區視網膜脈絡膜組織隨鞏膜組織向外凹陷,凹陷區域內視網膜神經上皮層薄變及缺損,神經上皮層間呈囊樣變化伴淺脫離
例2 例1之兄,29歲。因自幼雙眼視力差與例1同時來我院就診。眼科檢查,雙眼眼位正位,無眼球震顫。右眼視力0.1,?7.50 DS/+4.00 DC×90°=0.1;左眼視力0.05,?4.00 DS/+3.00 DC×85°=0.1。右眼眼壓19 mmHg,左眼眼壓20 mmHg。雙眼瞳孔對光反射遲鈍,其余眼前節檢查正常。雙眼視盤邊界清楚,顏色基本正常,C/D=0.3;視網膜平復,黃斑區可見約1.0 DD大小的類圓形缺損區(圖2A);其邊界清晰,凹陷深,中間可見灰白色鞏膜組織,脈絡膜毛細血管缺失。OCT檢查,雙眼黃斑缺損區視網膜脈絡膜組織隨鞏膜組織局限性向外凹陷,凹陷區域內視網膜神經上皮層變薄,部分缺損,神經上皮層間囊樣改變,其下見淺脫離(圖2B)。基因檢查:患者攜帶下列雜合突變:(1)CEP290 c. 3573+3A>G;(2)CEP290 c. 366A>G,p. K122K。診斷:雙眼先天性黃斑缺損(色素型)。
討論 先天性黃斑缺損多為常染色體顯性遺傳,少數為隱性遺傳,亦存在散發病例[1]。臨床表現為患眼中心視力差,注視不佳或斜視,眼底異常改變;病灶可位于黃斑或黃斑附近,表面平坦或輕中度凹陷,其大小、形狀和顏色呈不規則變化[2]。OCT檢查可反映黃斑缺損區組織結構的變化[3]。依據鞏膜暴露的程度及色素的多少本病可分為色素型、無色素型以及合并血管異常型[4]。本文報道的2例患者均可見黃斑區結構缺失伴色素沉著。根據OCT檢查并結合基因檢測結果,符合先天性黃斑缺損(色素型)診斷。2例患者的父母無臨床表現,但未行基因檢測,其發病的遺傳學特征有待進一步研究。
本病需與以下疾病鑒別:(1)Stargardt病:主要特征為黃斑區出現圓形或橢圓形的邊界清晰的黃色病灶,呈“牛眼”樣表現,病灶周圍及后極部可見黃色斑點[5];(2)脈絡膜缺損:脈絡膜局限性缺損常伴有其它眼部異常改變,如虹膜缺損、視神經發育不良或缺損、黃斑缺損等[6];(3)彌漫性脈絡膜營養障礙:后極部出現脈絡膜萎縮,繼而向視網膜周邊部緩慢進展,最終脈絡膜呈彌漫性萎縮,鞏膜暴露,無黃斑結構缺損;(4)中心性暈輪狀視網膜脈絡膜萎縮[7]:雙眼呈對稱性、邊界清晰的脈絡膜視網膜萎縮,萎縮區一般小于1.0 DD,無黃斑結構缺損。本文報道的2例患者眼底無“牛眼”樣改變及黃色斑點以及無虹膜、視神經缺損,可資鑒別。