引用本文: 林柳燕, 郝小波. 以黃斑囊樣水腫首診的Usher綜合征一例. 中華眼底病雜志, 2017, 33(6): 647-648. doi: 10.3760/cma.j.issn.1005-1015.2017.06.026 復制
患兒女,5歲。因左眼視物不清2年于2016年11月來我院就診。患兒足月順產,無吸氧史或窒息史;否認家族遺傳病史。患兒2歲時發現耳聾(具體檢查情況不詳),一直配戴助聽器,對答基本流利。2015年家長發現患兒雙眼視力差在外院就診。眼科檢查:右眼視力0.4,左眼視力0.01;眼前節檢查未見異常。因患兒年幼眼底檢查欠配合,未行眼底彩色照相。考慮屈光不正、弱視。配鏡及進行弱視治療,效果不佳。光相干斷層掃描(OCT)檢查,雙眼黃斑神經上皮層間多數無反射小囊腔。未給予處理。2016年2月復查,右眼視力0.3,左眼視力0.01。OCT檢查,雙眼黃斑神經上皮層間多數無反射囊腔;黃斑視網膜厚度增厚。考慮雙眼黃斑囊樣水腫(CME)。2~9月給予右眼2次、左眼3次玻璃體腔注射雷珠單抗0.03 ml治療。末次治療后復查,右眼矯正視力0.6,左眼矯正視力0.02。OCT檢查,右眼CME明顯消退,層間細小囊腔;左眼CME無變化(圖1)。家長為求中醫藥治療來我院就診。眼科檢查:右眼視力0.6,左眼視力0.02。眼底中周部視網膜可疑色素樣改變(圖2)。熒光素眼底血管造影(FFA)檢查,雙眼中周部視網膜呈斑駁樣透見熒光;晚期黃斑囊樣熒光素積存(圖3)。考慮視網膜色素變性(RP)。視網膜電圖(ERG)檢查,雙眼呈熄滅改變,震蕩電位a、b波低平消失。初步診斷:Usher綜合征Ⅱ型。基因檢查:pcdh15基因存在c.4970_4971、c.3045_3046insTGTG、c.91+1G>A 3個復合雜合變異。其中,c.4970_4971缺失,導致氨基酸改變P.S1657Ffs*31(移碼突變),其父親該位點雜合變異;c.3045_3046insTGTG導致氨基酸改變P.M1016c fs*17(移碼突變),其父親該位點雜合變異;c.91+1G>A導致氨基酸改變splicing(剪接突變),其母親該位點雜合變異。FCN2基因存在1個雜合突變,c1065_1067del缺失,導致氨基酸改變p.355_356del(缺失),患兒母親該位點雜合變異。最后診斷:Usher綜合征Ⅱ型;雙眼CME。
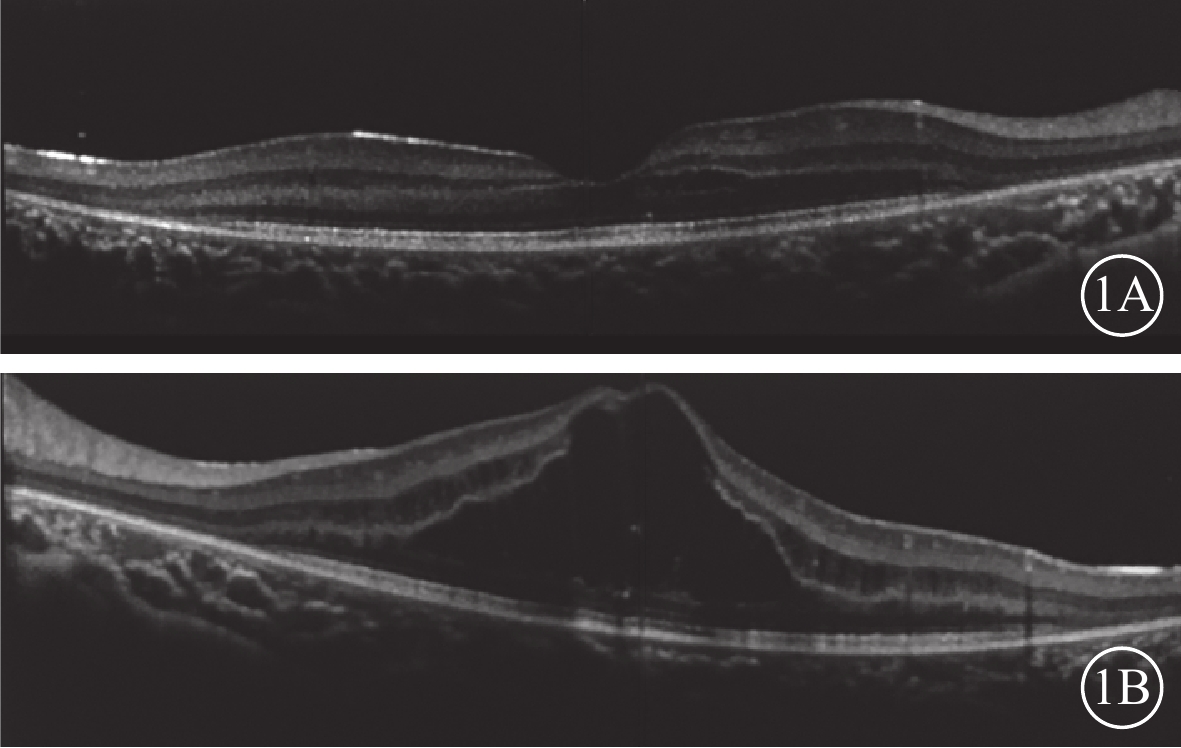 圖1
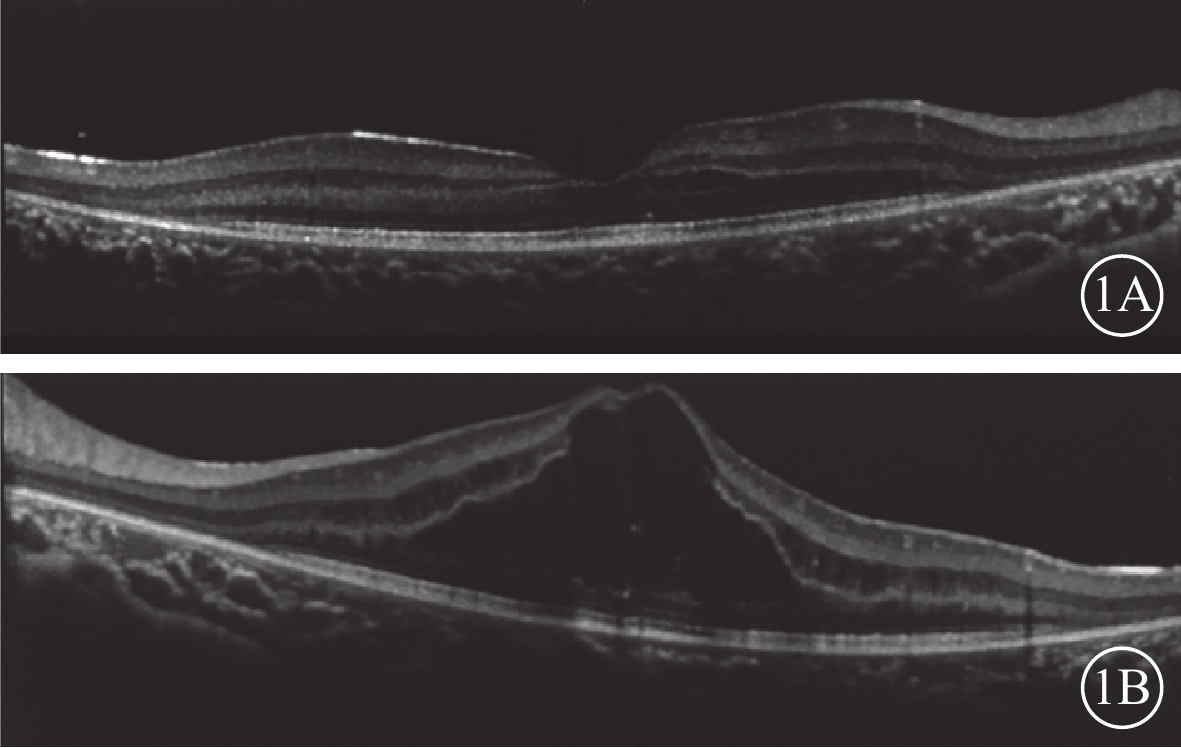
雙眼OCT像。1A. 右眼;1B. 左眼。雙眼黃斑鼻側神經上皮層內無反射小囊腔;左眼黃斑神經上皮層內無反射囊腔,層間視網膜劈裂
圖1
雙眼OCT像。1A. 右眼;1B. 左眼。雙眼黃斑鼻側神經上皮層內無反射小囊腔;左眼黃斑神經上皮層內無反射囊腔,層間視網膜劈裂
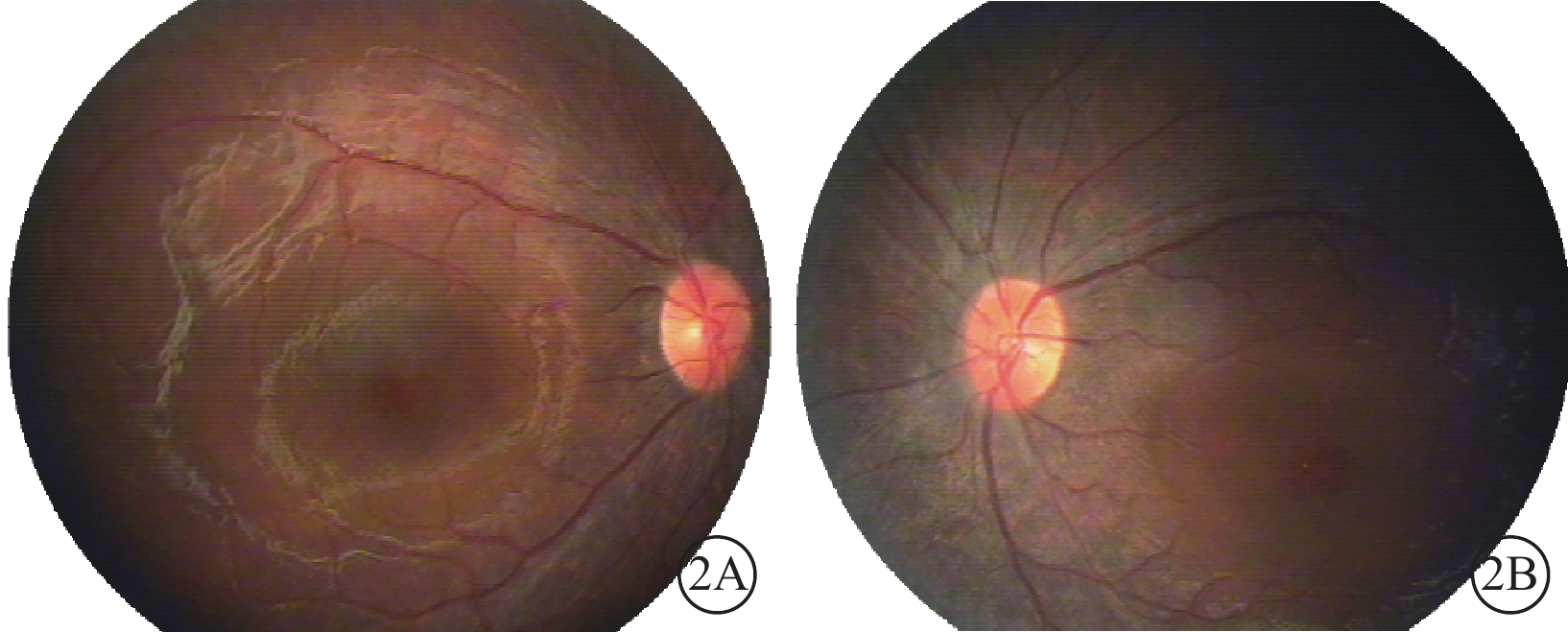 圖2
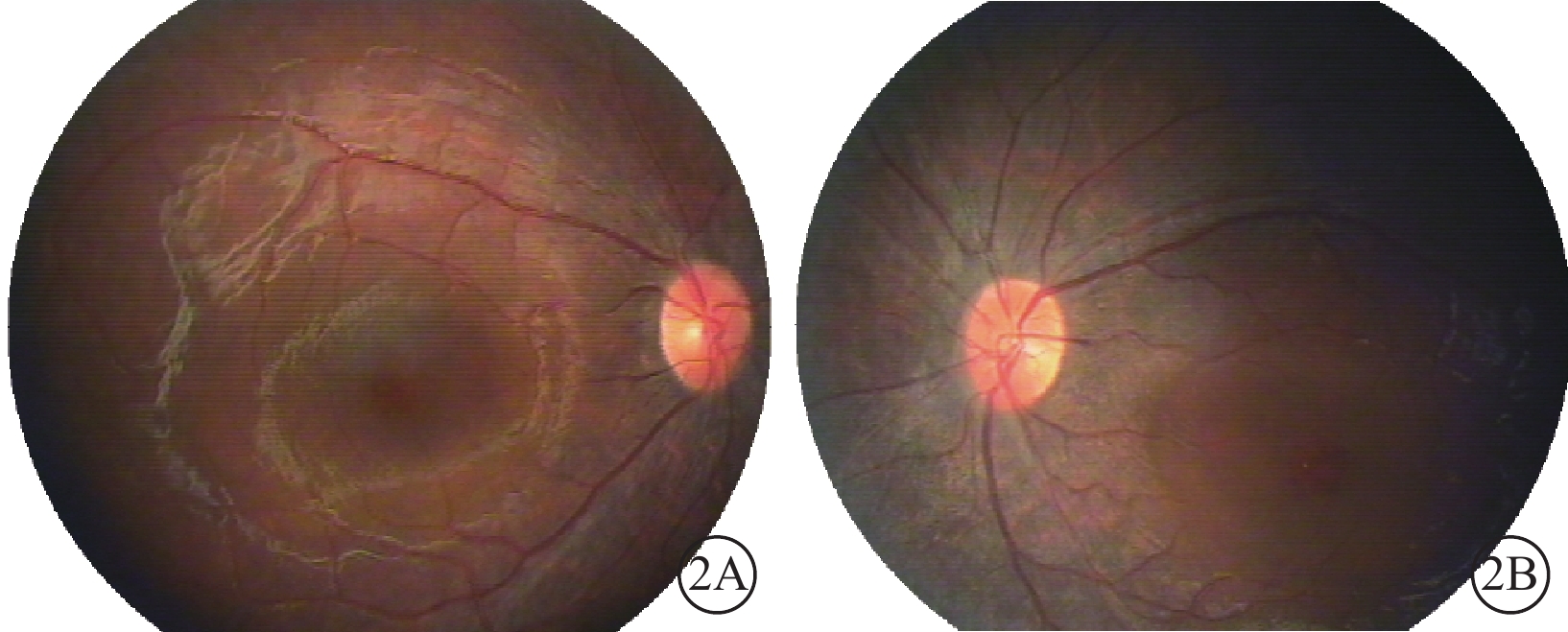
雙眼彩色眼底像。2A. 右眼;2B. 左眼。雙眼中周部視網膜可疑色素樣改變
圖2
雙眼彩色眼底像。2A. 右眼;2B. 左眼。雙眼中周部視網膜可疑色素樣改變
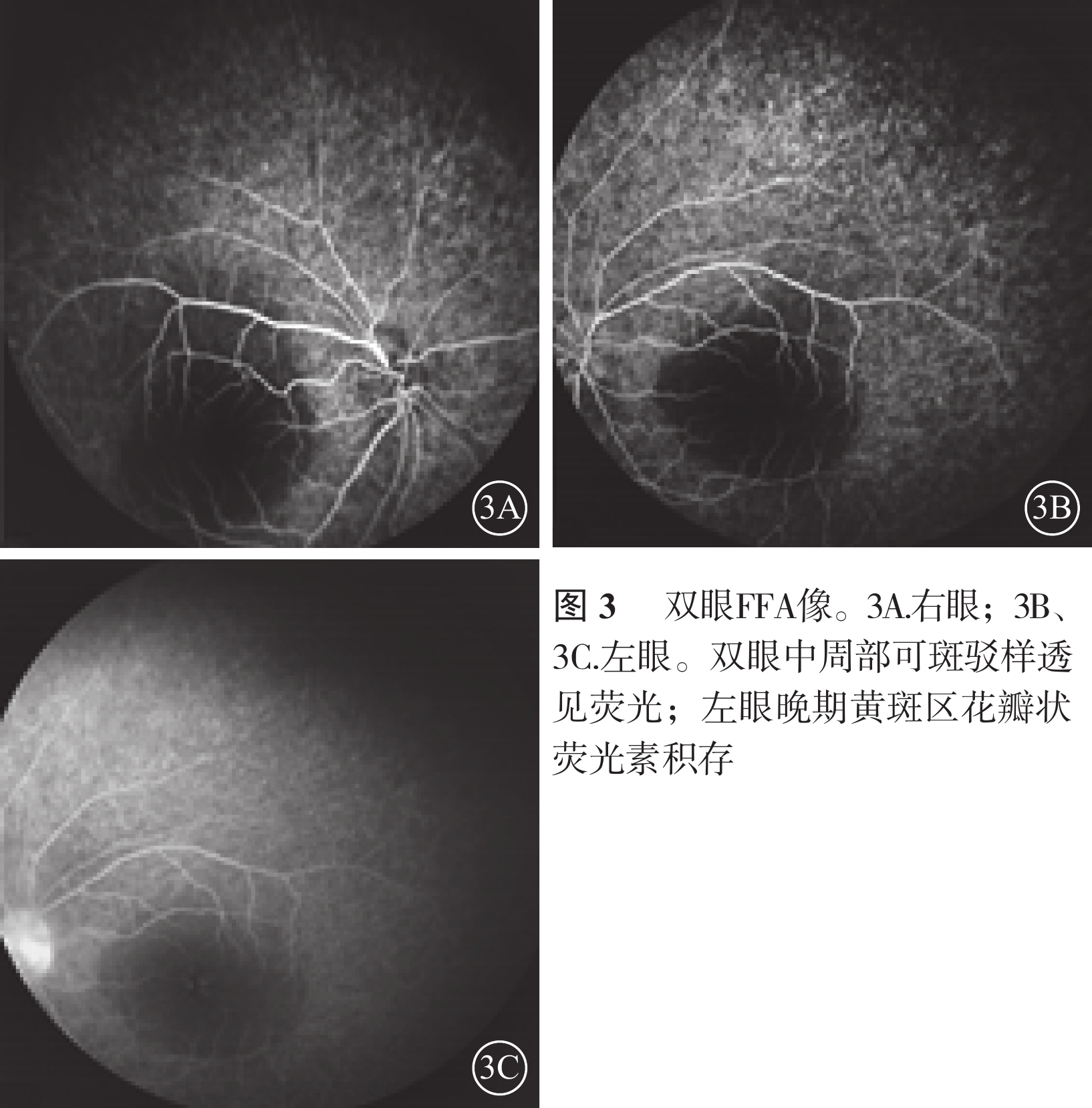 圖3
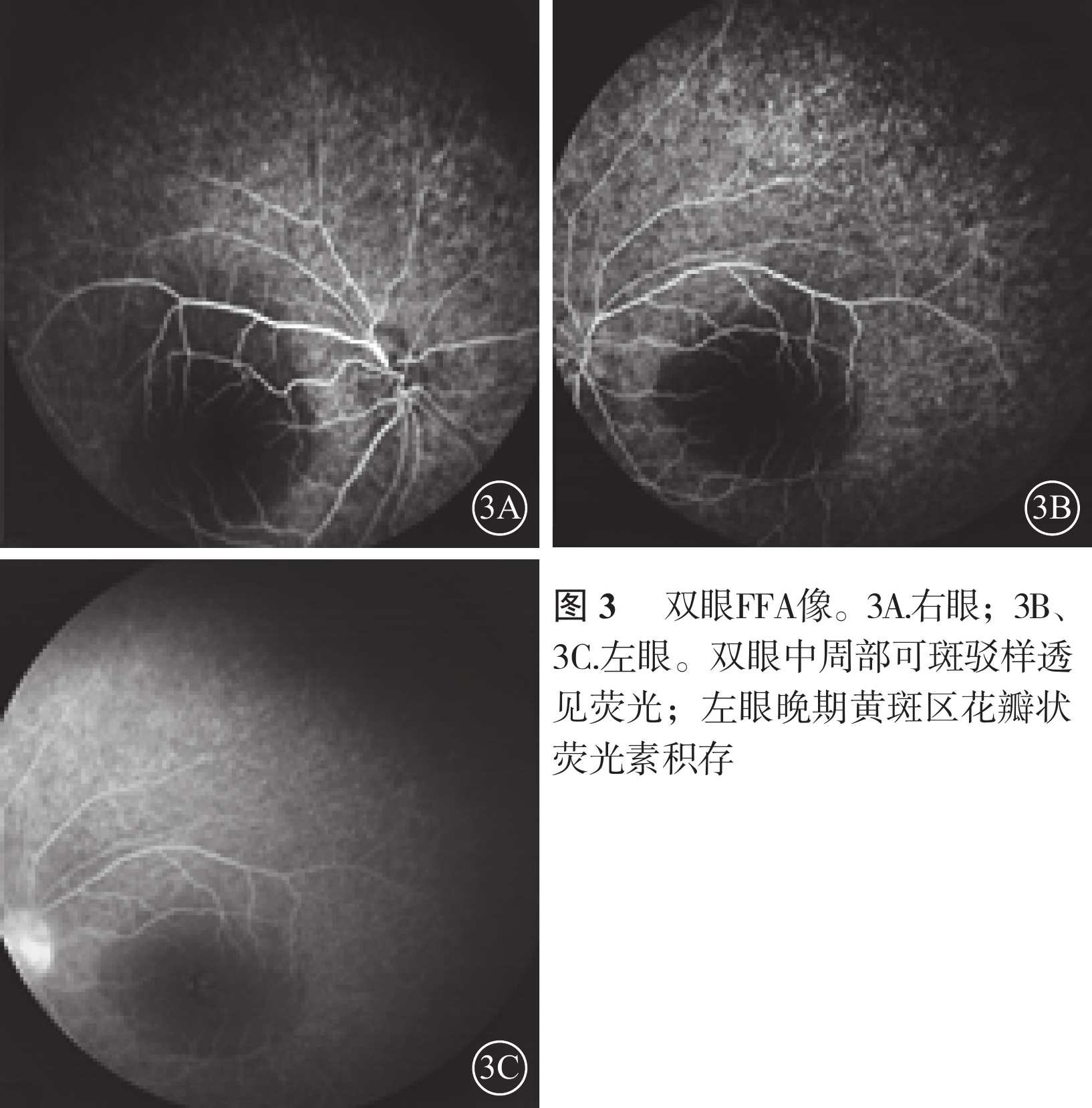
雙眼FFA像。3A. 右眼;3B、3C. 左眼。雙眼中周部可斑駁樣透見熒光;左眼晚期黃斑區花瓣狀熒光素積存
圖3
雙眼FFA像。3A. 右眼;3B、3C. 左眼。雙眼中周部可斑駁樣透見熒光;左眼晚期黃斑區花瓣狀熒光素積存
討論 Usher綜合征是一種以先天性感音神經性耳聾和漸進性RP為特征的常染色體隱性遺傳病[1]。臨床上根據聽力和前庭功能受累情況將其分為三型:Ⅰ型:先天性重深度感音神經性聾,前庭反應消失;Ⅱ型:先天性中重度感音神經性聾,前庭反應正常;Ⅲ型:進行性感音神經性聾,前庭反應正常[2]。本例患兒根據基因檢查結果及上述疾病分型特點,可明確診斷為Usher綜合征Ⅱ型。
導致本例患兒誤診的原因我們分析其原因為發病時患兒年齡小,無法配合完成多項功能檢查;多次OCT檢查以CME為主要表現;眼底彩色照相未見典型骨細胞樣色素沉著。進一步FFA檢查發現視網膜中周部斑駁樣透見熒光,從而考慮RP可能。提示臨床上以聽力障礙合并視力差就診的患兒,應考慮本病的可能。
RP合并CME的原因可能為視網膜色素上皮(RPE)細胞萎縮性改變,導致RPE泵功能受損,使視網膜向脈絡膜泵出液體能力減弱,最終導致CME發生[3]。也有學者認為疾病早期視桿細胞病變,造成內層視網膜缺血缺氧,血管內皮生長因子(VEGF)增加,從而表現為視網膜血管滲漏及CME形成[4]。
抗VEGF藥物治療是主要通過拮抗作用抑制新生血管生成、降低血管通透性、調控血視網膜屏障通透性,從而達到促進視網膜內滲液吸收和改善黃斑水腫的目的[5]。本例患兒行抗VEGF藥物治療后,右眼視力提高,CME改善;左眼視力及CME均無改善,治療效果差。可能與抗VEGF藥物對橢圓體帶完整的CME治療具有一定療效有關[6]。但是針對一些特發性的黃斑水腫仍是治療難題。本例患兒RP以視網膜血管變細,視網膜血運差,感光細胞萎縮為特點,抗VEGF藥物是否會增加內層細胞缺血臨床上仍需要長期觀察隨訪和進一步研究。
患兒女,5歲。因左眼視物不清2年于2016年11月來我院就診。患兒足月順產,無吸氧史或窒息史;否認家族遺傳病史。患兒2歲時發現耳聾(具體檢查情況不詳),一直配戴助聽器,對答基本流利。2015年家長發現患兒雙眼視力差在外院就診。眼科檢查:右眼視力0.4,左眼視力0.01;眼前節檢查未見異常。因患兒年幼眼底檢查欠配合,未行眼底彩色照相。考慮屈光不正、弱視。配鏡及進行弱視治療,效果不佳。光相干斷層掃描(OCT)檢查,雙眼黃斑神經上皮層間多數無反射小囊腔。未給予處理。2016年2月復查,右眼視力0.3,左眼視力0.01。OCT檢查,雙眼黃斑神經上皮層間多數無反射囊腔;黃斑視網膜厚度增厚。考慮雙眼黃斑囊樣水腫(CME)。2~9月給予右眼2次、左眼3次玻璃體腔注射雷珠單抗0.03 ml治療。末次治療后復查,右眼矯正視力0.6,左眼矯正視力0.02。OCT檢查,右眼CME明顯消退,層間細小囊腔;左眼CME無變化(圖1)。家長為求中醫藥治療來我院就診。眼科檢查:右眼視力0.6,左眼視力0.02。眼底中周部視網膜可疑色素樣改變(圖2)。熒光素眼底血管造影(FFA)檢查,雙眼中周部視網膜呈斑駁樣透見熒光;晚期黃斑囊樣熒光素積存(圖3)。考慮視網膜色素變性(RP)。視網膜電圖(ERG)檢查,雙眼呈熄滅改變,震蕩電位a、b波低平消失。初步診斷:Usher綜合征Ⅱ型。基因檢查:pcdh15基因存在c.4970_4971、c.3045_3046insTGTG、c.91+1G>A 3個復合雜合變異。其中,c.4970_4971缺失,導致氨基酸改變P.S1657Ffs*31(移碼突變),其父親該位點雜合變異;c.3045_3046insTGTG導致氨基酸改變P.M1016c fs*17(移碼突變),其父親該位點雜合變異;c.91+1G>A導致氨基酸改變splicing(剪接突變),其母親該位點雜合變異。FCN2基因存在1個雜合突變,c1065_1067del缺失,導致氨基酸改變p.355_356del(缺失),患兒母親該位點雜合變異。最后診斷:Usher綜合征Ⅱ型;雙眼CME。
圖1
雙眼OCT像。1A. 右眼;1B. 左眼。雙眼黃斑鼻側神經上皮層內無反射小囊腔;左眼黃斑神經上皮層內無反射囊腔,層間視網膜劈裂
圖2
雙眼彩色眼底像。2A. 右眼;2B. 左眼。雙眼中周部視網膜可疑色素樣改變
圖3
雙眼FFA像。3A. 右眼;3B、3C. 左眼。雙眼中周部可斑駁樣透見熒光;左眼晚期黃斑區花瓣狀熒光素積存
討論 Usher綜合征是一種以先天性感音神經性耳聾和漸進性RP為特征的常染色體隱性遺傳病[1]。臨床上根據聽力和前庭功能受累情況將其分為三型:Ⅰ型:先天性重深度感音神經性聾,前庭反應消失;Ⅱ型:先天性中重度感音神經性聾,前庭反應正常;Ⅲ型:進行性感音神經性聾,前庭反應正常[2]。本例患兒根據基因檢查結果及上述疾病分型特點,可明確診斷為Usher綜合征Ⅱ型。
導致本例患兒誤診的原因我們分析其原因為發病時患兒年齡小,無法配合完成多項功能檢查;多次OCT檢查以CME為主要表現;眼底彩色照相未見典型骨細胞樣色素沉著。進一步FFA檢查發現視網膜中周部斑駁樣透見熒光,從而考慮RP可能。提示臨床上以聽力障礙合并視力差就診的患兒,應考慮本病的可能。
RP合并CME的原因可能為視網膜色素上皮(RPE)細胞萎縮性改變,導致RPE泵功能受損,使視網膜向脈絡膜泵出液體能力減弱,最終導致CME發生[3]。也有學者認為疾病早期視桿細胞病變,造成內層視網膜缺血缺氧,血管內皮生長因子(VEGF)增加,從而表現為視網膜血管滲漏及CME形成[4]。
抗VEGF藥物治療是主要通過拮抗作用抑制新生血管生成、降低血管通透性、調控血視網膜屏障通透性,從而達到促進視網膜內滲液吸收和改善黃斑水腫的目的[5]。本例患兒行抗VEGF藥物治療后,右眼視力提高,CME改善;左眼視力及CME均無改善,治療效果差。可能與抗VEGF藥物對橢圓體帶完整的CME治療具有一定療效有關[6]。但是針對一些特發性的黃斑水腫仍是治療難題。本例患兒RP以視網膜血管變細,視網膜血運差,感光細胞萎縮為特點,抗VEGF藥物是否會增加內層細胞缺血臨床上仍需要長期觀察隨訪和進一步研究。