引用本文: 孔繁強, 周樹民, 陳松. 基質金屬蛋白酶-9調控CD73自視網膜色素上皮細胞膜表面脫落機制的初步研究. 中華眼底病雜志, 2017, 33(5): 518-522. doi: 10.3760/cma.j.issn.1005-1015.2017.05.018 復制
三磷酸腺苷(ATP)及其代謝產物不僅是重要的能量代謝物質,同時也具有顯著的免疫調控作用[1, 2]。被大量釋放至細胞外的ATP可加重局部炎癥反應[3-5]。但當ATP被分解代謝為腺苷(adenosine)后則轉變為高效的免疫抑制劑[6-8]。ATP至腺苷的轉化過程對局部免疫反應具有重要的調控作用。CD73是控制腺苷產生的限速酶,諸多細胞可通過其表面的CD73促進腺苷的產生,發揮免疫抑制作用[9-11]。視網膜色素上皮(RPE)細胞是眼底組織中唯一表達CD73的細胞;當局部炎癥發生時RPE細胞膜表面CD73含量急劇降低,不能在局部形成高腺苷濃度,對T細胞的抑制功能減弱而促進炎癥發生[12]。但CD73在RPE細胞膜表面的含量變化是由何種機制所調控尚未明了。經檢索文獻及對CD73的結構分析發現,磷脂酶C(PLC)和基質金屬蛋白酶(MMP)可能參與了CD73自RPE細胞表面的脫落過程。PLC通過水解CD73所錨定的糖基磷酯酰肌醇(GPI)而使CD73脫落;MMP則通過水解CD73中潛在的MMP識別位點Lys547-Phe548間的肽鍵而致CD73的脫落。我們通過比較PLC抑制劑、MMP抑制劑對CD73脫落的影響,初步探討CD73自RPE細胞膜表面可能的脫落機制。現將結果報道如下。
1 材料和方法
人RPE細胞株(APRE-19)及含野生型(Wt)人CD73表達序列的pCDNA載體pcDNA-wtcd73(本實驗室保存)。PLC抑制劑ET-18-OCH3(美國Sigma公司)。廣譜MMP抑制劑ONO-4817、半選擇性MMP-2/9抑制劑Ⅴ、選擇性MMP-2抑制劑ARP100、選擇性MMP-9抑制劑Ⅰ、MMP-2 siRNA、MMP-9 siRNA及無標靶siRNA(NT-siRNA)(美國Santa Cruz公司)。藻紅蛋白(PE)標記的抗RPE65、異硫氰酸熒光素標記的抗CD73抗體(美國eBioscience公司)。轉染試劑lipofectamine 2000、脂多糖(LPS)、腫瘤壞死因子(TNF)-α及定點突變試劑盒(美國Invitrogen公司)。
誘導CD73自RPE細胞膜表面脫落及酶抑制劑干預。于24孔細胞培養板中體外培養APRE-19至80%融合,預留6副孔細胞,不進行任何處理作為無干預對照組。剩余細胞均加入50 ng/ml LPS和200 ng/ml TNF-α進行干預。依據酶抑制劑干預方式的不同進一步分為PLC抑制劑組、MMP-2抑制劑、MMP-2/9抑制劑、MMP-9抑制劑組,每組均為6副孔。其中,PLC抑制劑ET-18-OCH3終濃度為30.0 μmol/L、廣譜MMP抑制劑ONO-4817終濃度為5.0 μmol/L、半選擇性MMP-2/9抑制劑V終濃度為5.0 μmol/L、選擇性MMP-2抑制劑ARP100終濃度為3.0 μmol/L、選擇性MMP-9抑制劑CTK8G1150終濃度分別為0.5、1.0、2.0、5.0μmol/L[13-15]。未加入酶抑制劑的細胞作為溶媒組。48 h后每組取3孔細胞以熒光標記抗體染色,流式細胞儀檢測RPE細胞膜表面CD73含量。另3孔細胞采用定量聚合酶鏈反應檢測細胞膜表面CD73 mRNA的相對表達量。以磷酸甘油醛脫氫酶為內參照,其表達量以與無干預對照組的比值表示。
siRNA下調MMP-9表達抑制CD73脫落。脂質體試劑介導NT-siRNA、MMP-2 siRNA及MMP-9 siRNA轉染體外培養的ARPE-19,同時設立無轉染的對照細胞。48 h后分別裂解細胞,蛋白質免疫印跡法(Western blot)檢測RPE細胞內MMP-2、MMP-9含量。經不同siRNA轉染或未轉染的RPE細胞在48 h后給予LPS及TNF-α聯合干預。干預48 h后行抗體染色及流式細胞儀檢測,以直方圖顯示有、無PE-抗CD73抗體染色的熒光曲線表示RPE細胞膜表面CD73的含量。
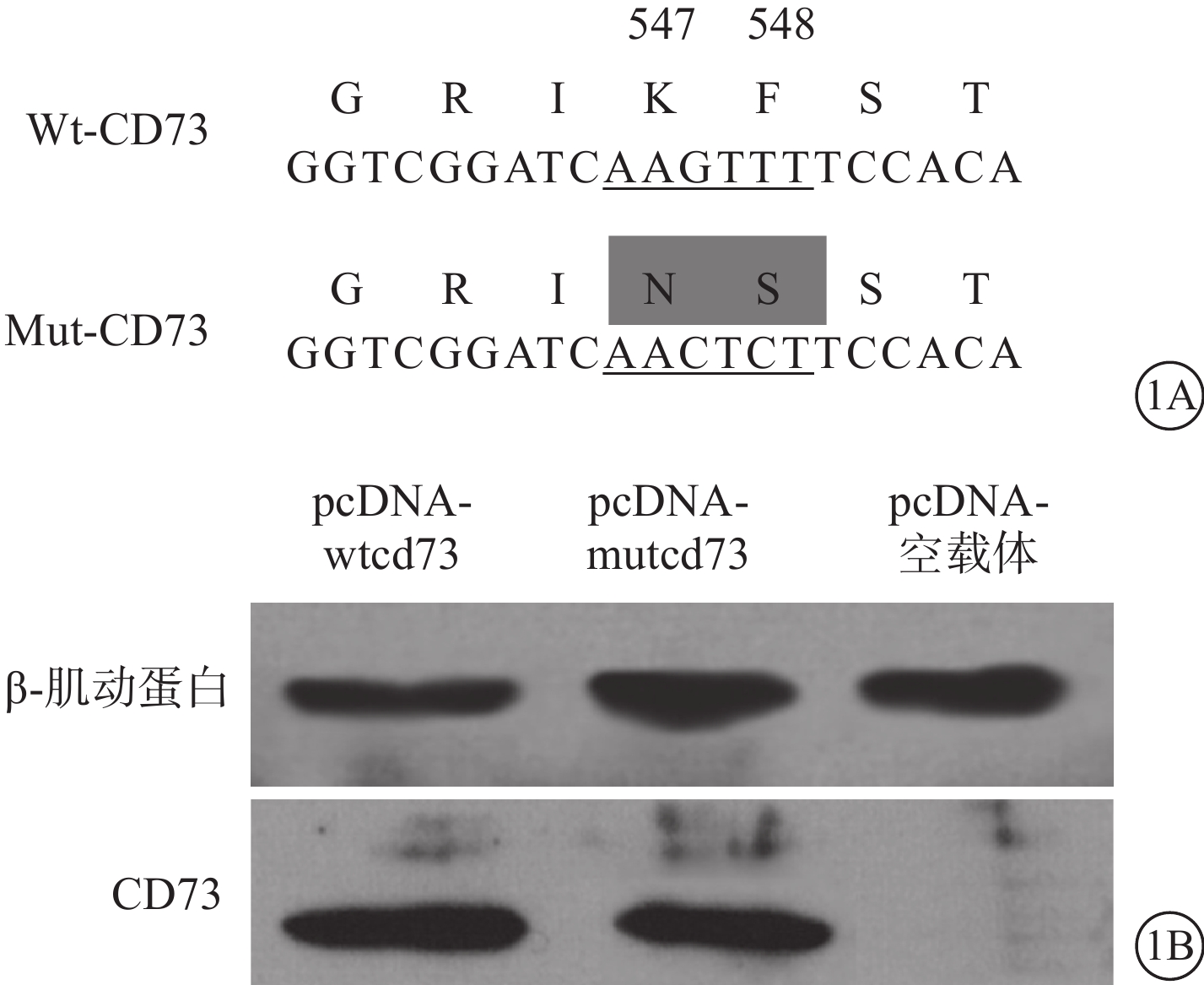
Wt、突變型(Mut)-CD73在CD73-/- RPE細胞中的表達。通過定點突變技術將重組質粒pcDNA-wtcd73中編碼Lys547及Phe548的序列突變為Asn547及Ser548的編碼序列,獲得可表達Mut-CD73的重組質粒pcDNA-mutcd73(圖1A);兩種重組質粒分別在CD73-/-RPE細胞中進行表達,Western blot檢測并獲得驗證(圖1B)CD73-/- RPE細胞分離自CD73-/- 小鼠。經2~3代傳代培養后分別接受脂質體介導的重組pcDNA-wtcd73、pcDNA-mutcd73質粒及pcDNA空載體轉染,轉染于24孔細胞培養板中進行。 48 h后各組另取3孔細胞合并后Western blot檢測外源CD73在CD73-/- RPE細胞中的表達。剩余細胞給予LPS及TNF-α聯合干預,同時給予或不給予選擇性MMP-9抑制劑,每種干預方式設立3副孔。繼續培養48 h后流式細胞儀檢測RPE細胞膜表面CD73含量。
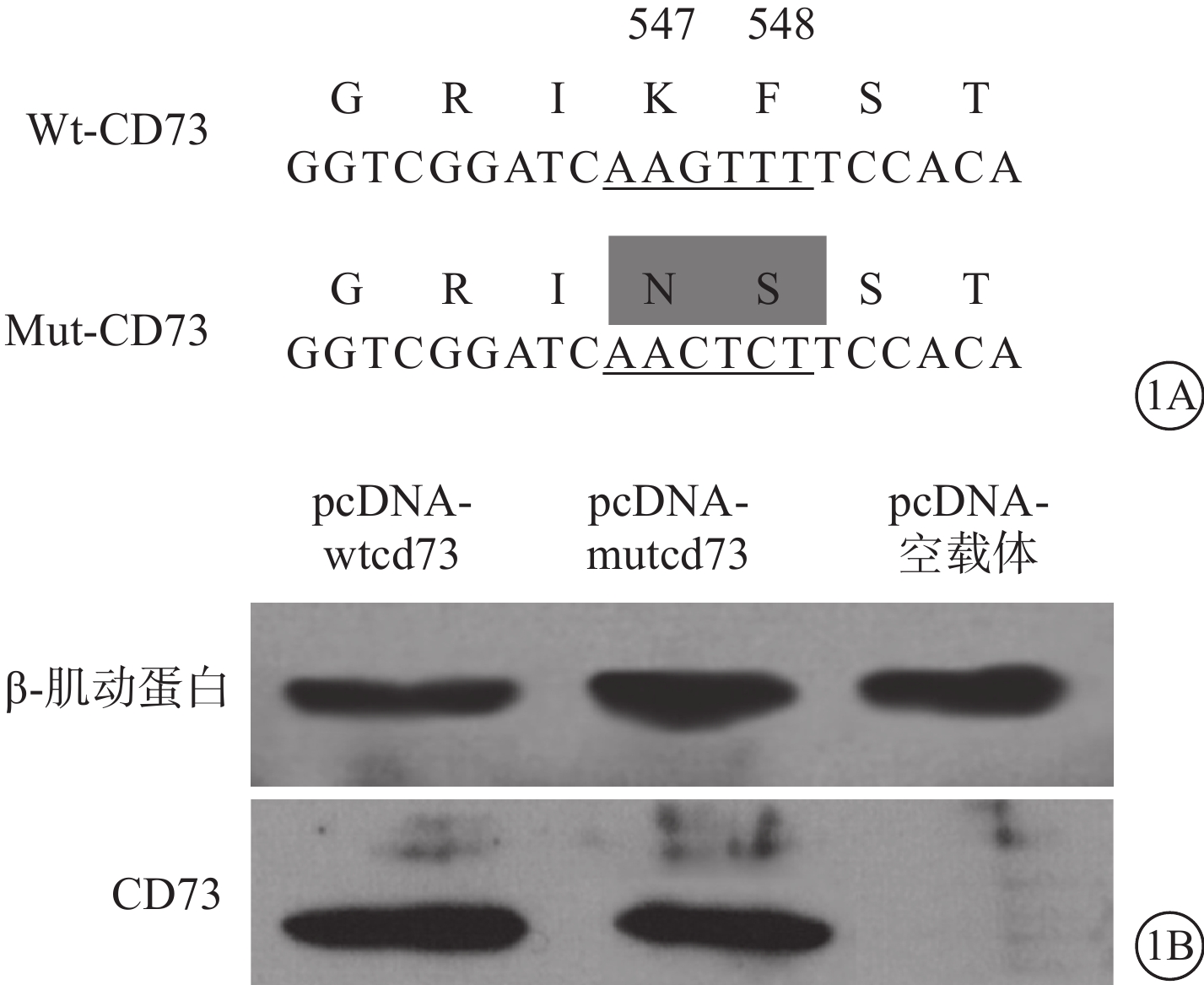 圖1
Wt、Mut-CD73序列及在CD73-/- RPE細胞中表達示意圖。1A. Wt、Mut-CD73基因編碼序列及對應的氨基酸;1B. 電泳圖,Wt、Mut-CD73基因在CD73-/- RPE細胞中的表達結果
圖1
Wt、Mut-CD73序列及在CD73-/- RPE細胞中表達示意圖。1A. Wt、Mut-CD73基因編碼序列及對應的氨基酸;1B. 電泳圖,Wt、Mut-CD73基因在CD73-/- RPE細胞中的表達結果
采用SPSS 16.0統計軟件進行統計學分析處理。實驗數據行方差分析,兩兩比較行SNK-q檢驗。P<0.05為差異有統計學意義。
2 結果
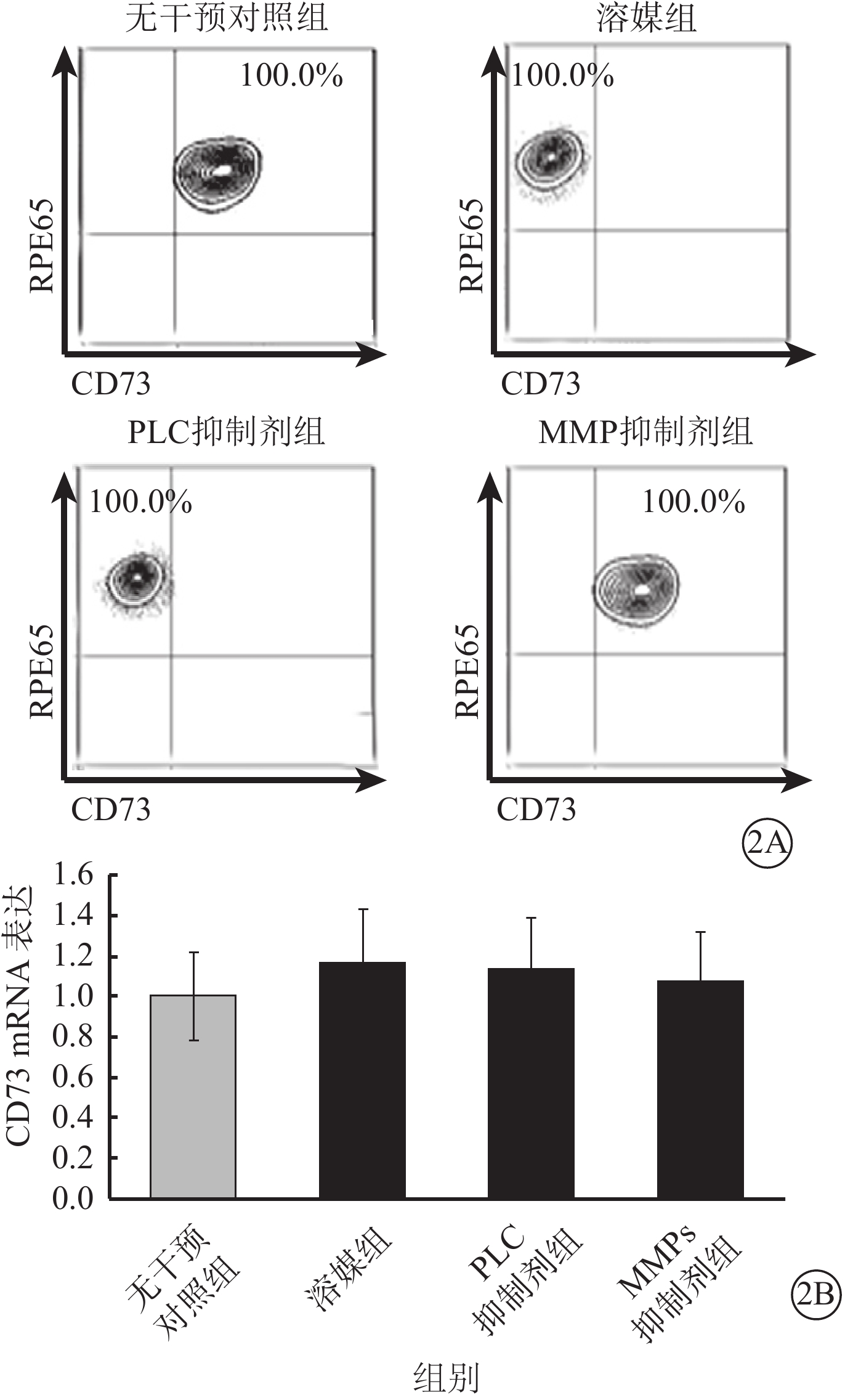
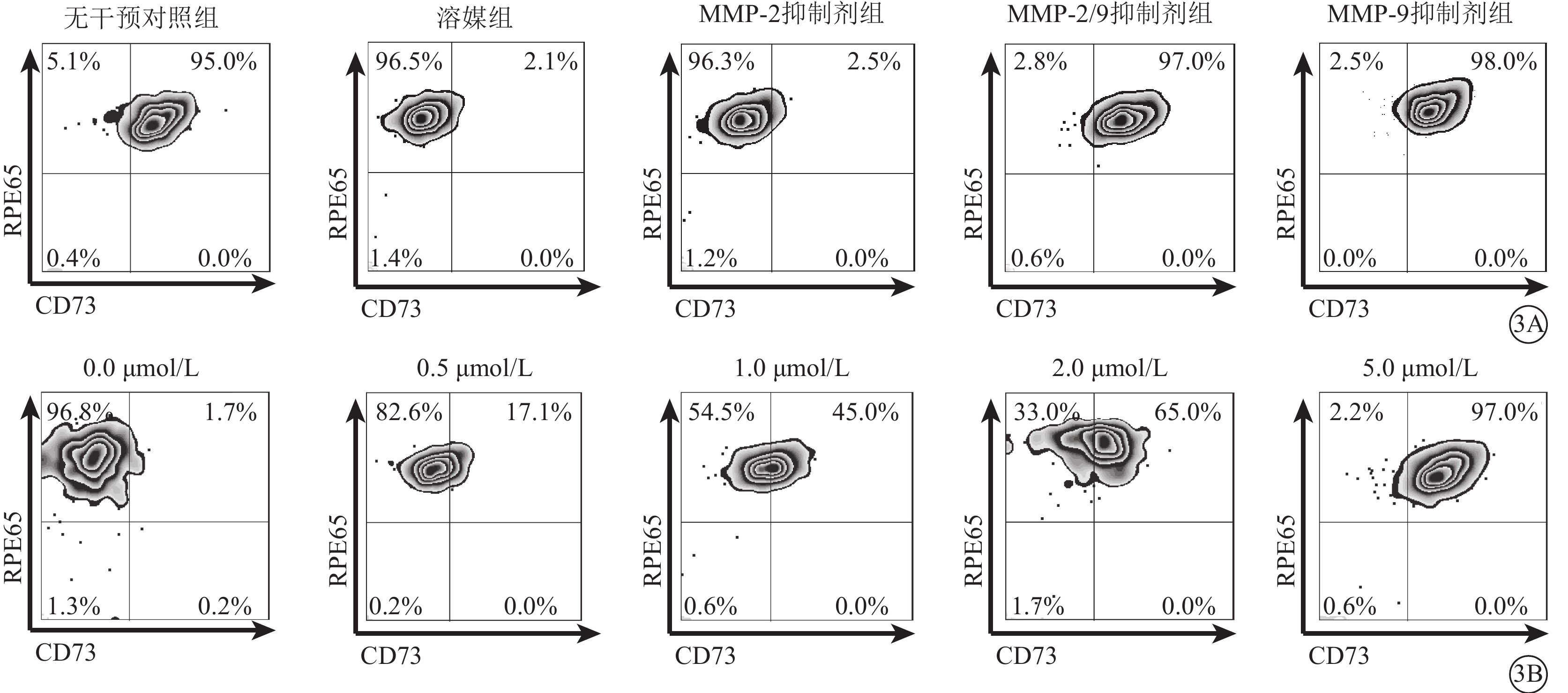
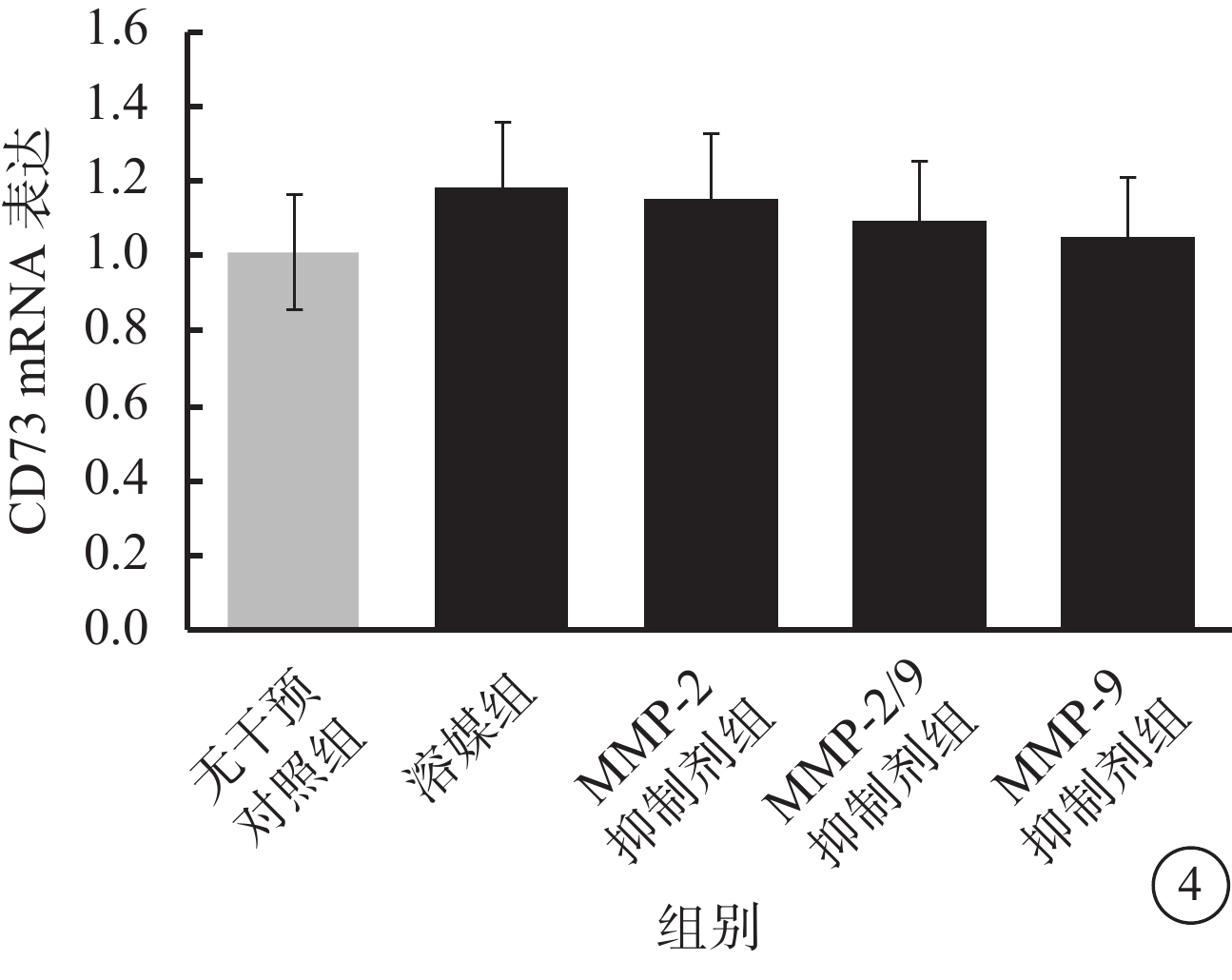
流式細胞儀檢測發現,LPS及TNF-α聯合干預可使RPE細胞膜表面CD73陽性轉變為陰性;此過程可被廣譜MMP抑制劑所拮抗,對PLC抑制劑則無反應(圖2A);CD73 mRNA在RPE細胞膜中的表達并未因干預方式的不同而發生顯著性變化(F=0.545,P>0.05)(圖2B)。半選擇性MMP-2/9抑制劑及選擇性MMP-9抑制劑可有效阻斷LPS、TNF-α誘導的RPE細胞膜表面CD73陰性化,選擇性MMP-2抑制劑則無類似作用(圖3A);MMP-9抑制劑對CD73陰性化阻斷作用呈明顯劑量依賴關系(圖3B)。不同干預方式均未顯著影響CD73 mRNA在RPE細胞膜中的表達(F=0.665,P>0.05)(圖4)。
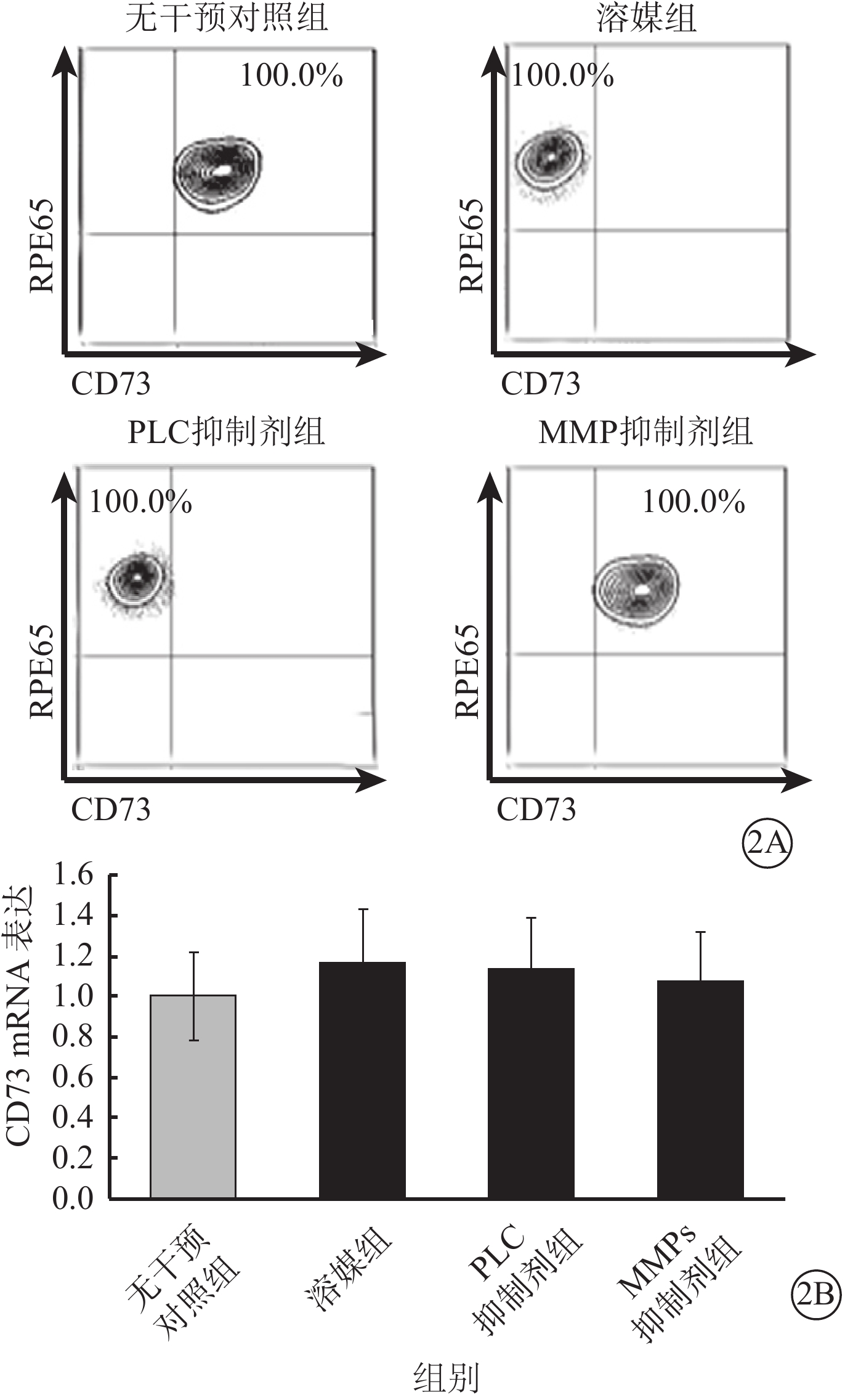 圖2
不同干預方式對RPE細胞膜中CD73 mRNA表達影響。2A.流式細胞圖;2B.RPE細胞膜中CD73 mRNA表達結果
圖2
不同干預方式對RPE細胞膜中CD73 mRNA表達影響。2A.流式細胞圖;2B.RPE細胞膜中CD73 mRNA表達結果
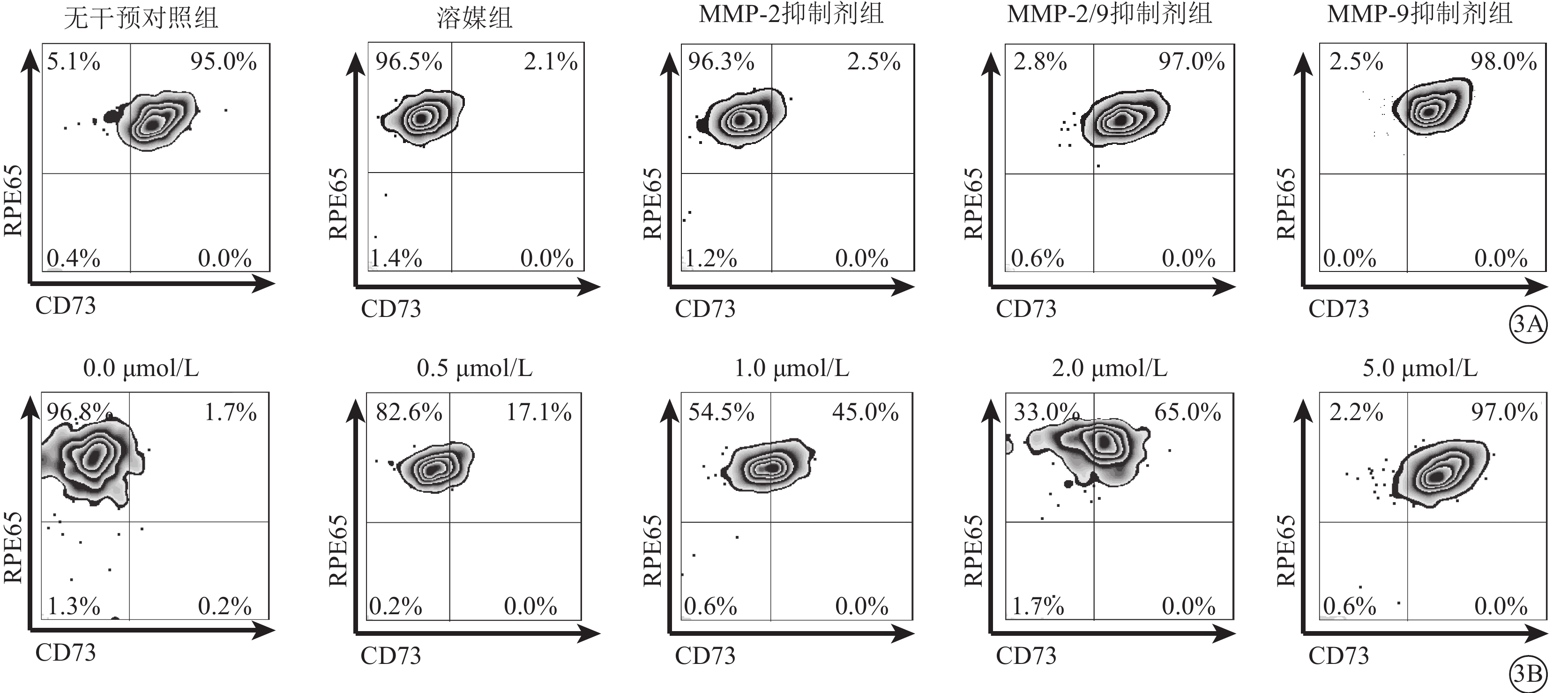 圖3
選擇性MMP-2、MMP-9抑制劑和半選擇性MMP-2/9抑制劑對RPE細胞膜中CD73 mRNA表達影響。3A. 流式細胞圖,選擇性MMP-2、MMP-9抑制劑和半選擇性MMP-2/9抑制劑;3B. 流式細胞圖,不同濃度MMP-9抑制劑
圖3
選擇性MMP-2、MMP-9抑制劑和半選擇性MMP-2/9抑制劑對RPE細胞膜中CD73 mRNA表達影響。3A. 流式細胞圖,選擇性MMP-2、MMP-9抑制劑和半選擇性MMP-2/9抑制劑;3B. 流式細胞圖,不同濃度MMP-9抑制劑
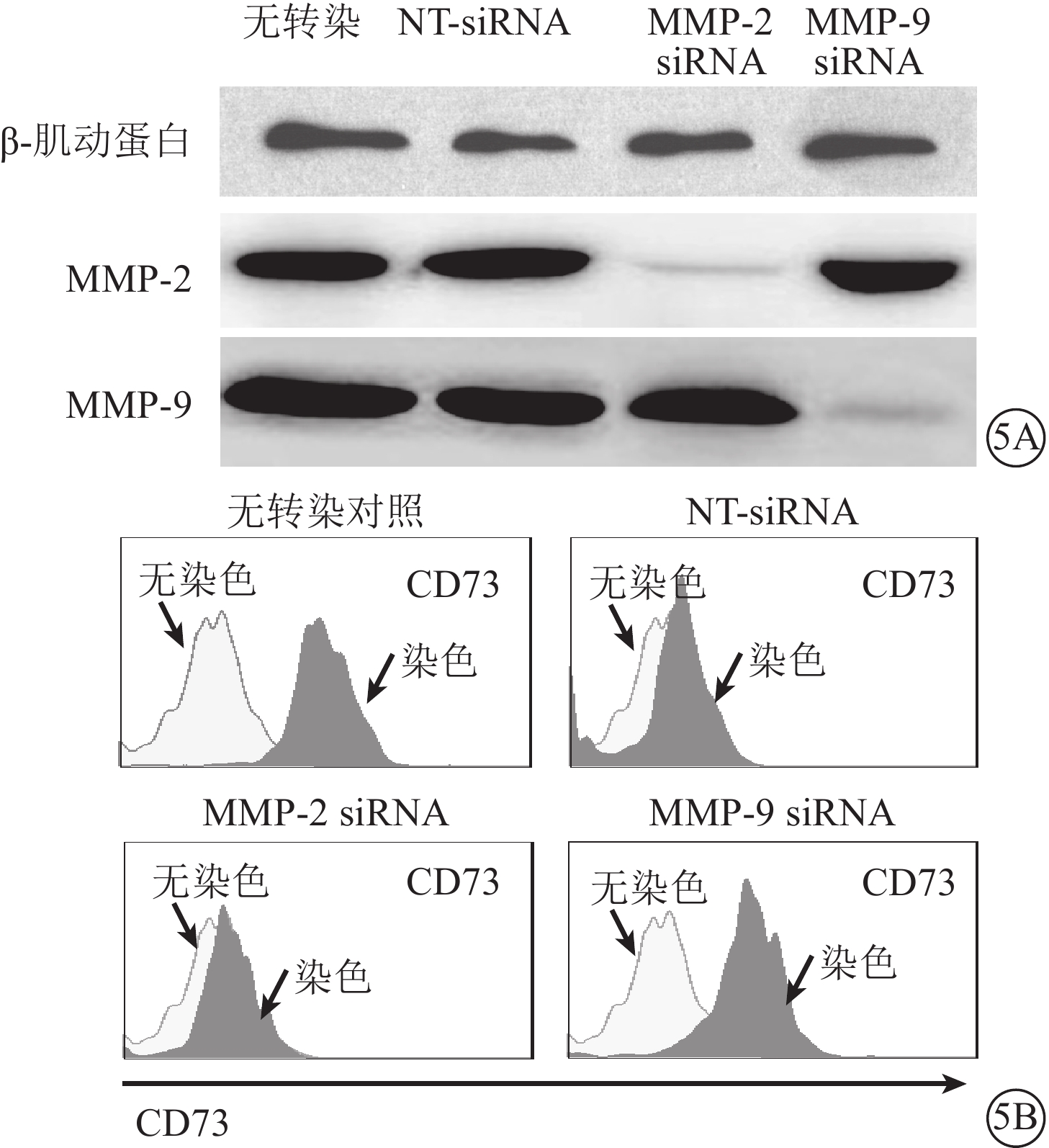
Western blot檢測發現,轉染48 h后,MMP-2、MMP-9 siRNA可特異性下調對應靶分子在RPE細胞中的表達(圖5A)。LPS、TNF-α所誘導的CD73自RPE細胞膜表面脫落可有效被MMP-9 siRNA轉染(圖5B),但不能被MMP-2 siRNA轉染拮抗。
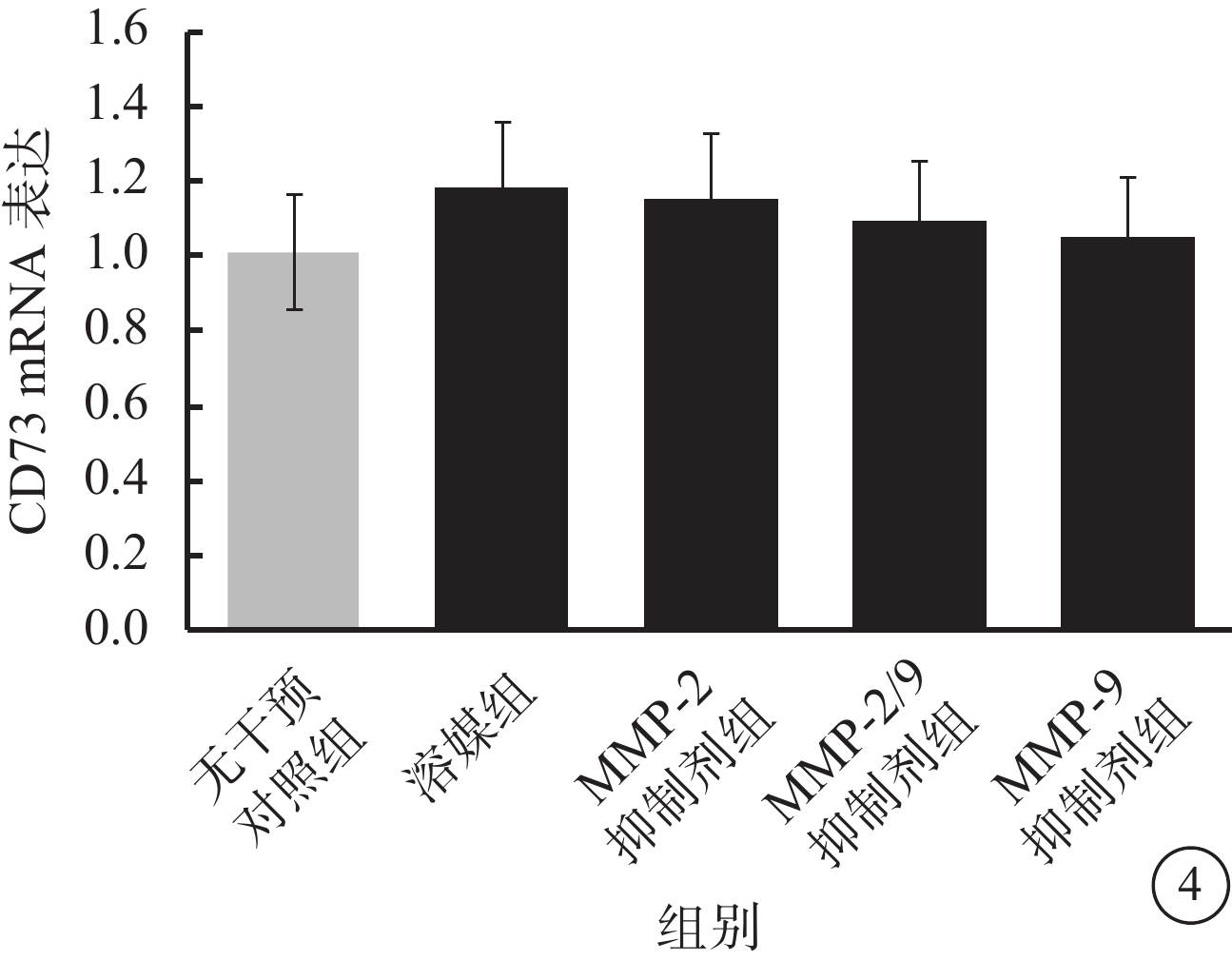 圖4
REF細胞膜中CD73 mRNA表達結果
圖4
REF細胞膜中CD73 mRNA表達結果
流式細胞儀檢測發現,LPS、TNF-α誘導后,Wt-CD73可以自CD73-/-RPE細胞膜表面脫落;Mut-CD73則不脫落(圖6)。
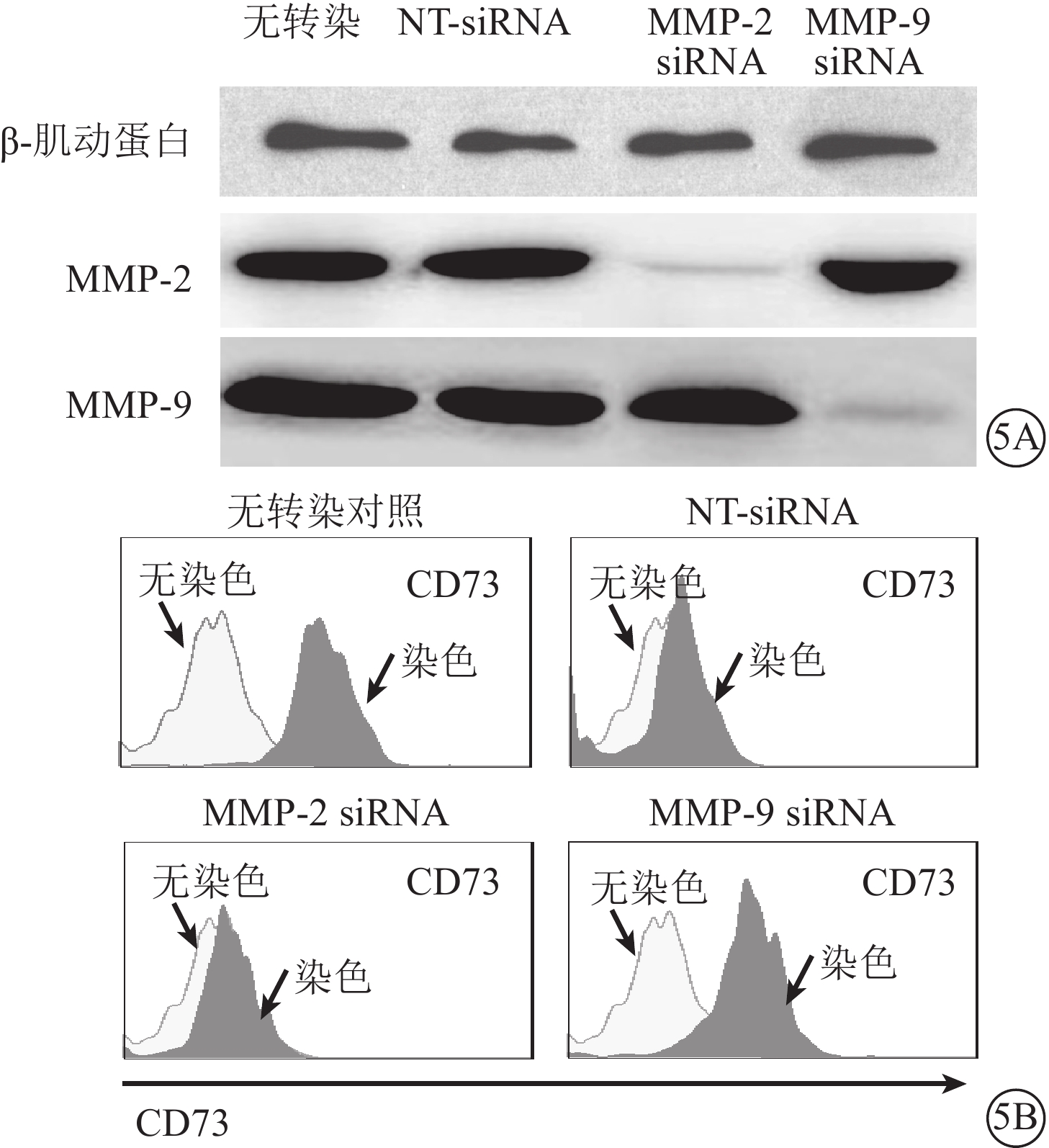 圖5
MMP-9 siRNA下調靶基因表達,抑制CD73自RPE細胞膜表面的脫落。5A. 電泳圖,MMP-2、MMP-9 siRNA選擇性下調靶基因的表達;5B. 流式細胞圖,MMP-9 siRNA轉染抑制CD73自RPE細胞膜表面脫落結果
圖5
MMP-9 siRNA下調靶基因表達,抑制CD73自RPE細胞膜表面的脫落。5A. 電泳圖,MMP-2、MMP-9 siRNA選擇性下調靶基因的表達;5B. 流式細胞圖,MMP-9 siRNA轉染抑制CD73自RPE細胞膜表面脫落結果
3 討論
除對眼底組織的支持、營養、保護等作用外,RPE細胞對局部免疫反應的調節作用近年來備受關注。生理條件下RPE細胞主要發揮免疫抑制作用,促使眼底組織處于免疫豁免狀態[16, 17]。當某些病理因素存在時,RPE細胞又可表現出某些免疫促進作用,如遞呈抗原刺激T細胞的激活[18],分泌多種趨化因子募集炎性細胞[19]。其中,RPE細胞膜表面CD73在生理、病理情況下的顯著變化也參與了其對免疫調節作用的轉變。因此,了解CD73如何由生理狀況下在RPE細胞膜表面的陽性存在而轉變為病理情況下的陰性,有助于了解某些眼底炎癥的發生,并為其治療提供潛在的靶點。
CD73本身并不具備跨膜結構域,其羧基端與嵌合在細胞膜磷脂雙層中的GPI相結合而錨定于細胞膜上。兩種酶可潛在催化CD73自RPE細胞表面膜上脫落,一種是PLC,水解GPI與細胞膜磷脂雙層的結合;另一種是蛋白酶,如MMP,水解CD73中的肽鍵而導致CD73的脫落。
本研究結果顯示,廣譜MMP抑制劑可有效阻止LPS、TNF-α所誘發的CD73陰性化。但此作用并非通過抑制CD73 mRNA的表達實現,由此可確定MMP通過水解CD73使其自RPE細胞膜表面脫落。既往有關MMP在RPE細胞膜中的表達研究主要集中于MMP-2、MMP- 9[20-22]。因此,本研究也將MMP-2、MMP-9作為重點。無論是選擇性抑制劑干預,還是siRNA轉染的結果均顯示是MMP-9介導CD73自RPE細胞膜表面的水解。通過對CD73中MMP-9的識別位點進行分析,發現在CD73子中MMP-9的潛在酶解位點為Lys547-Phe548間的肽鍵,并且應用定點突變技術對Wt-CD73編碼序列進行改造后證實,Lys547 - Phe548位點突變后的CD73可在CD73-/-RPE細胞膜的表面持續存在而不發生脫落。為此,本研究結果證實MMP-9通過水解CD73中的Lys547-Phe548位點而促使CD73自RPE細胞膜表面脫落。
三磷酸腺苷(ATP)及其代謝產物不僅是重要的能量代謝物質,同時也具有顯著的免疫調控作用[1, 2]。被大量釋放至細胞外的ATP可加重局部炎癥反應[3-5]。但當ATP被分解代謝為腺苷(adenosine)后則轉變為高效的免疫抑制劑[6-8]。ATP至腺苷的轉化過程對局部免疫反應具有重要的調控作用。CD73是控制腺苷產生的限速酶,諸多細胞可通過其表面的CD73促進腺苷的產生,發揮免疫抑制作用[9-11]。視網膜色素上皮(RPE)細胞是眼底組織中唯一表達CD73的細胞;當局部炎癥發生時RPE細胞膜表面CD73含量急劇降低,不能在局部形成高腺苷濃度,對T細胞的抑制功能減弱而促進炎癥發生[12]。但CD73在RPE細胞膜表面的含量變化是由何種機制所調控尚未明了。經檢索文獻及對CD73的結構分析發現,磷脂酶C(PLC)和基質金屬蛋白酶(MMP)可能參與了CD73自RPE細胞表面的脫落過程。PLC通過水解CD73所錨定的糖基磷酯酰肌醇(GPI)而使CD73脫落;MMP則通過水解CD73中潛在的MMP識別位點Lys547-Phe548間的肽鍵而致CD73的脫落。我們通過比較PLC抑制劑、MMP抑制劑對CD73脫落的影響,初步探討CD73自RPE細胞膜表面可能的脫落機制。現將結果報道如下。
1 材料和方法
人RPE細胞株(APRE-19)及含野生型(Wt)人CD73表達序列的pCDNA載體pcDNA-wtcd73(本實驗室保存)。PLC抑制劑ET-18-OCH3(美國Sigma公司)。廣譜MMP抑制劑ONO-4817、半選擇性MMP-2/9抑制劑Ⅴ、選擇性MMP-2抑制劑ARP100、選擇性MMP-9抑制劑Ⅰ、MMP-2 siRNA、MMP-9 siRNA及無標靶siRNA(NT-siRNA)(美國Santa Cruz公司)。藻紅蛋白(PE)標記的抗RPE65、異硫氰酸熒光素標記的抗CD73抗體(美國eBioscience公司)。轉染試劑lipofectamine 2000、脂多糖(LPS)、腫瘤壞死因子(TNF)-α及定點突變試劑盒(美國Invitrogen公司)。
誘導CD73自RPE細胞膜表面脫落及酶抑制劑干預。于24孔細胞培養板中體外培養APRE-19至80%融合,預留6副孔細胞,不進行任何處理作為無干預對照組。剩余細胞均加入50 ng/ml LPS和200 ng/ml TNF-α進行干預。依據酶抑制劑干預方式的不同進一步分為PLC抑制劑組、MMP-2抑制劑、MMP-2/9抑制劑、MMP-9抑制劑組,每組均為6副孔。其中,PLC抑制劑ET-18-OCH3終濃度為30.0 μmol/L、廣譜MMP抑制劑ONO-4817終濃度為5.0 μmol/L、半選擇性MMP-2/9抑制劑V終濃度為5.0 μmol/L、選擇性MMP-2抑制劑ARP100終濃度為3.0 μmol/L、選擇性MMP-9抑制劑CTK8G1150終濃度分別為0.5、1.0、2.0、5.0μmol/L[13-15]。未加入酶抑制劑的細胞作為溶媒組。48 h后每組取3孔細胞以熒光標記抗體染色,流式細胞儀檢測RPE細胞膜表面CD73含量。另3孔細胞采用定量聚合酶鏈反應檢測細胞膜表面CD73 mRNA的相對表達量。以磷酸甘油醛脫氫酶為內參照,其表達量以與無干預對照組的比值表示。
siRNA下調MMP-9表達抑制CD73脫落。脂質體試劑介導NT-siRNA、MMP-2 siRNA及MMP-9 siRNA轉染體外培養的ARPE-19,同時設立無轉染的對照細胞。48 h后分別裂解細胞,蛋白質免疫印跡法(Western blot)檢測RPE細胞內MMP-2、MMP-9含量。經不同siRNA轉染或未轉染的RPE細胞在48 h后給予LPS及TNF-α聯合干預。干預48 h后行抗體染色及流式細胞儀檢測,以直方圖顯示有、無PE-抗CD73抗體染色的熒光曲線表示RPE細胞膜表面CD73的含量。
Wt、突變型(Mut)-CD73在CD73-/- RPE細胞中的表達。通過定點突變技術將重組質粒pcDNA-wtcd73中編碼Lys547及Phe548的序列突變為Asn547及Ser548的編碼序列,獲得可表達Mut-CD73的重組質粒pcDNA-mutcd73(圖1A);兩種重組質粒分別在CD73-/-RPE細胞中進行表達,Western blot檢測并獲得驗證(圖1B)CD73-/- RPE細胞分離自CD73-/- 小鼠。經2~3代傳代培養后分別接受脂質體介導的重組pcDNA-wtcd73、pcDNA-mutcd73質粒及pcDNA空載體轉染,轉染于24孔細胞培養板中進行。 48 h后各組另取3孔細胞合并后Western blot檢測外源CD73在CD73-/- RPE細胞中的表達。剩余細胞給予LPS及TNF-α聯合干預,同時給予或不給予選擇性MMP-9抑制劑,每種干預方式設立3副孔。繼續培養48 h后流式細胞儀檢測RPE細胞膜表面CD73含量。
圖1
Wt、Mut-CD73序列及在CD73-/- RPE細胞中表達示意圖。1A. Wt、Mut-CD73基因編碼序列及對應的氨基酸;1B. 電泳圖,Wt、Mut-CD73基因在CD73-/- RPE細胞中的表達結果
采用SPSS 16.0統計軟件進行統計學分析處理。實驗數據行方差分析,兩兩比較行SNK-q檢驗。P<0.05為差異有統計學意義。
2 結果
流式細胞儀檢測發現,LPS及TNF-α聯合干預可使RPE細胞膜表面CD73陽性轉變為陰性;此過程可被廣譜MMP抑制劑所拮抗,對PLC抑制劑則無反應(圖2A);CD73 mRNA在RPE細胞膜中的表達并未因干預方式的不同而發生顯著性變化(F=0.545,P>0.05)(圖2B)。半選擇性MMP-2/9抑制劑及選擇性MMP-9抑制劑可有效阻斷LPS、TNF-α誘導的RPE細胞膜表面CD73陰性化,選擇性MMP-2抑制劑則無類似作用(圖3A);MMP-9抑制劑對CD73陰性化阻斷作用呈明顯劑量依賴關系(圖3B)。不同干預方式均未顯著影響CD73 mRNA在RPE細胞膜中的表達(F=0.665,P>0.05)(圖4)。
圖2
不同干預方式對RPE細胞膜中CD73 mRNA表達影響。2A.流式細胞圖;2B.RPE細胞膜中CD73 mRNA表達結果
圖3
選擇性MMP-2、MMP-9抑制劑和半選擇性MMP-2/9抑制劑對RPE細胞膜中CD73 mRNA表達影響。3A. 流式細胞圖,選擇性MMP-2、MMP-9抑制劑和半選擇性MMP-2/9抑制劑;3B. 流式細胞圖,不同濃度MMP-9抑制劑
Western blot檢測發現,轉染48 h后,MMP-2、MMP-9 siRNA可特異性下調對應靶分子在RPE細胞中的表達(圖5A)。LPS、TNF-α所誘導的CD73自RPE細胞膜表面脫落可有效被MMP-9 siRNA轉染(圖5B),但不能被MMP-2 siRNA轉染拮抗。
圖4
REF細胞膜中CD73 mRNA表達結果
流式細胞儀檢測發現,LPS、TNF-α誘導后,Wt-CD73可以自CD73-/-RPE細胞膜表面脫落;Mut-CD73則不脫落(圖6)。
圖5
MMP-9 siRNA下調靶基因表達,抑制CD73自RPE細胞膜表面的脫落。5A. 電泳圖,MMP-2、MMP-9 siRNA選擇性下調靶基因的表達;5B. 流式細胞圖,MMP-9 siRNA轉染抑制CD73自RPE細胞膜表面脫落結果
3 討論
除對眼底組織的支持、營養、保護等作用外,RPE細胞對局部免疫反應的調節作用近年來備受關注。生理條件下RPE細胞主要發揮免疫抑制作用,促使眼底組織處于免疫豁免狀態[16, 17]。當某些病理因素存在時,RPE細胞又可表現出某些免疫促進作用,如遞呈抗原刺激T細胞的激活[18],分泌多種趨化因子募集炎性細胞[19]。其中,RPE細胞膜表面CD73在生理、病理情況下的顯著變化也參與了其對免疫調節作用的轉變。因此,了解CD73如何由生理狀況下在RPE細胞膜表面的陽性存在而轉變為病理情況下的陰性,有助于了解某些眼底炎癥的發生,并為其治療提供潛在的靶點。
CD73本身并不具備跨膜結構域,其羧基端與嵌合在細胞膜磷脂雙層中的GPI相結合而錨定于細胞膜上。兩種酶可潛在催化CD73自RPE細胞表面膜上脫落,一種是PLC,水解GPI與細胞膜磷脂雙層的結合;另一種是蛋白酶,如MMP,水解CD73中的肽鍵而導致CD73的脫落。
本研究結果顯示,廣譜MMP抑制劑可有效阻止LPS、TNF-α所誘發的CD73陰性化。但此作用并非通過抑制CD73 mRNA的表達實現,由此可確定MMP通過水解CD73使其自RPE細胞膜表面脫落。既往有關MMP在RPE細胞膜中的表達研究主要集中于MMP-2、MMP- 9[20-22]。因此,本研究也將MMP-2、MMP-9作為重點。無論是選擇性抑制劑干預,還是siRNA轉染的結果均顯示是MMP-9介導CD73自RPE細胞膜表面的水解。通過對CD73中MMP-9的識別位點進行分析,發現在CD73子中MMP-9的潛在酶解位點為Lys547-Phe548間的肽鍵,并且應用定點突變技術對Wt-CD73編碼序列進行改造后證實,Lys547 - Phe548位點突變后的CD73可在CD73-/-RPE細胞膜的表面持續存在而不發生脫落。為此,本研究結果證實MMP-9通過水解CD73中的Lys547-Phe548位點而促使CD73自RPE細胞膜表面脫落。