引用本文: 趙娜, 陸勤康, 王惠云, 童奇湖. 常染色體顯性遺傳視錐-視桿營養不良一家系的基因突變檢測. 中華眼底病雜志, 2016, 32(6): 637-639. doi: 10.3760/cma.j.issn.1005-1015.2016.06.017 復制
視錐-視桿營養不良(CORD)為高度臨床異質性和遺傳異質性眼病,以視錐細胞受損為主,伴不同程度的視桿細胞損傷[1-3]。目前, 已明確的CORD致病基因已有25個[1-3]。其中,常染色體隱性遺傳基因13個;常染色體顯性遺傳基因10個;X連鎖遺傳基因2個。本研究對1個南方漢族常染色體顯性遺傳CORD家系的視錐-視桿同源盒(CRX)基因突變譜進行了測序驗證。現將結果報道如下。
1 對象和方法
本研究經本院醫學倫理委員會審核批準。參與研究的所有個體均簽署知情同意書。
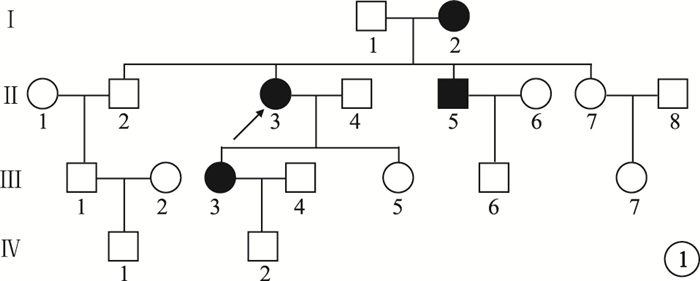
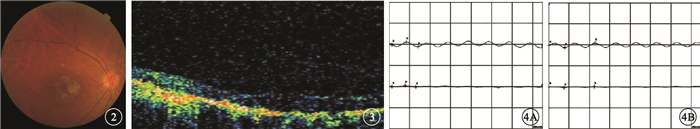
來自南方的1個4代19人漢族家系,其中CODR患者4例(圖 1)。對所有家系成員均行眼科檢查。先證者,女,52歲。10余年前雙眼開始視力進行性下降。右眼視力0.1,左眼視力0.15,均不能矯正。畏光但無眼球震顫;色覺輕微異常。眼底黃斑區2個視盤直徑(DD)×2 DD金箔樣反光,散在色素沉著,中心凹反光消失,伴黃色斑點;中周部視網膜可見骨細胞樣色素沉著,視網膜動脈血管變細(圖 2)。光相干斷層掃描(OCT)檢查可見黃斑區外層視網膜萎縮變薄(圖 3)。閃光視網膜電圖(FERG)檢查,暗適應b波振幅下降,視桿反應輕度下降;明適應a、b波振幅均明顯下降,視錐反應消失(圖 4)。熒光素眼底血管造影(FFA)檢查黃斑區呈強熒光,未見脈絡膜湮沒(圖 5)。家系其余3例患者中,男性1例,女性2例。年齡分別為75、48、32歲;發病年齡分別為40、39、32歲。視力0.05~0.4。均有畏光但無眼球震顫。色覺檢查,輕微異常、色盲、色弱各1例。眼底黃斑區1.0 DD×1.5 DD金箔樣反光,散在色素沉著1例;黃斑區色素沉著2例。中心凹反光消失,伴黃色斑點,視網膜色素沉著1例;中心凹反光不清2例,其中外周視網膜可見針尖樣骨細胞色素沉著1例。FERG檢查,視錐反應消失1例,明顯下降2例;視桿反應輕度下降2例,正常1例。其余家系成員檢查結果未見異常。依據臨床檢查結果和家系分析,結合國內外文獻[1-4],初步診斷為常染色體顯性遺傳CORD (CORD2)。
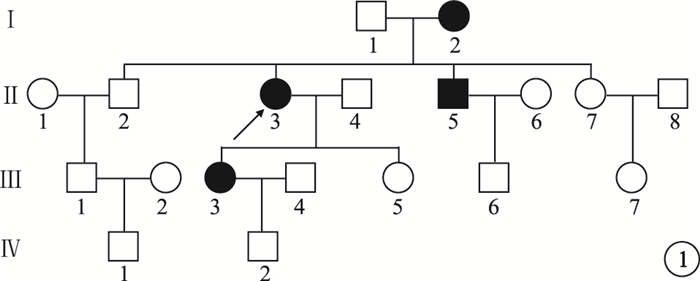 圖1
患者家系圖。■:男性患者;●:女性患者;↑:先證者;□:正常男性;○:正常女性
圖1
患者家系圖。■:男性患者;●:女性患者;↑:先證者;□:正常男性;○:正常女性
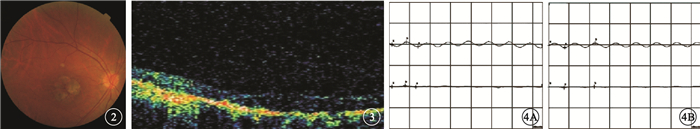 圖2
先證者右眼彩色眼底像。黃斑區不規則圓形黃色萎縮灶,暴露脈絡膜大血管,其間雜色素沉著,后極部見黃色斑點??圖 3??先證者右眼OCT像。黃斑區外層視網膜萎縮變薄??圖 4??先證者雙眼FERG像。4A.暗適應b波振幅下降,視桿反應輕度下降;4B.明適應a、b波振幅明顯下降,視錐反應消失
圖2
先證者右眼彩色眼底像。黃斑區不規則圓形黃色萎縮灶,暴露脈絡膜大血管,其間雜色素沉著,后極部見黃色斑點??圖 3??先證者右眼OCT像。黃斑區外層視網膜萎縮變薄??圖 4??先證者雙眼FERG像。4A.暗適應b波振幅下降,視桿反應輕度下降;4B.明適應a、b波振幅明顯下降,視錐反應消失
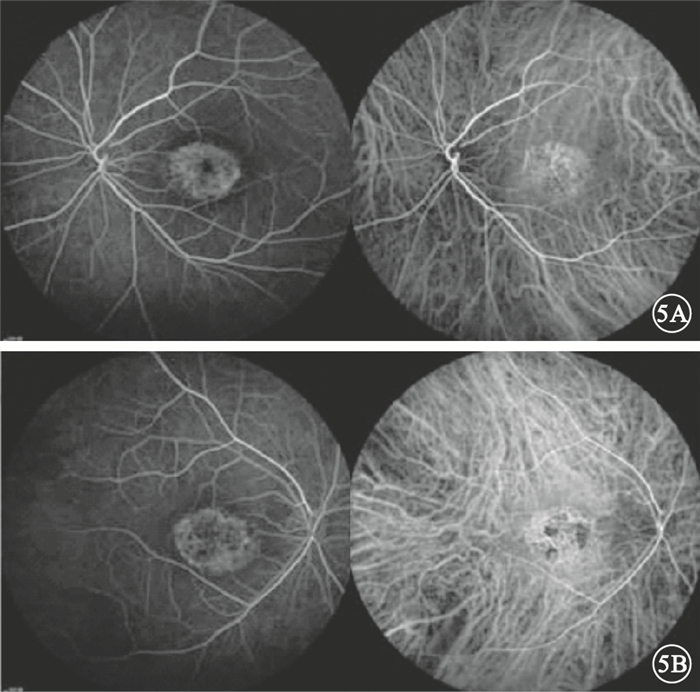
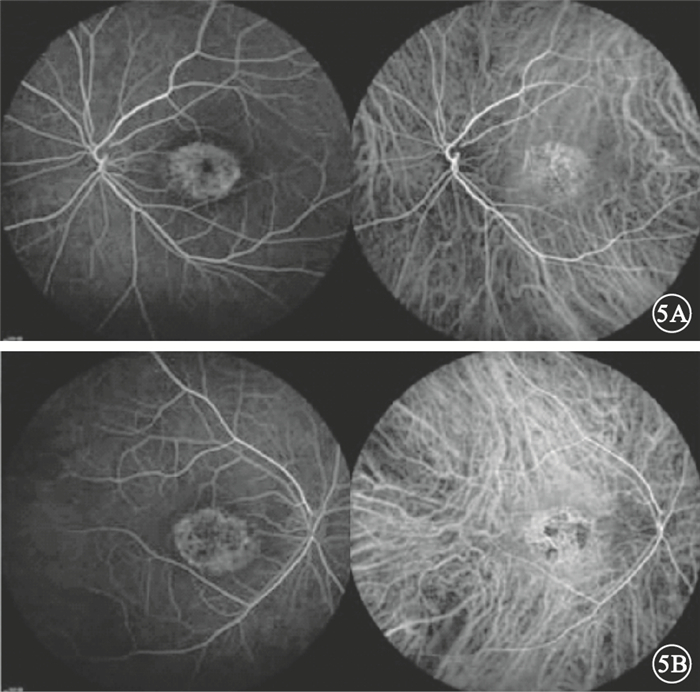 圖5
先證者雙眼FFA像。5A.左眼,早期黃斑區見強熒光,晚期呈透見熒光;5B.右眼,黃斑“靶的”狀強熒光,透見與遮蔽熒光相間,晚期色素上皮病變部位呈斑駁狀強熒光
圖5
先證者雙眼FFA像。5A.左眼,早期黃斑區見強熒光,晚期呈透見熒光;5B.右眼,黃斑“靶的”狀強熒光,透見與遮蔽熒光相間,晚期色素上皮病變部位呈斑駁狀強熒光
采集家系所有成員的外周血5 ml,乙二胺四乙酸抗凝。天根血液基因組DNA抽提試劑盒(北京天根生化科技有限公司)提取基因組DNA。應用Primer 5.0軟件設計CRX基因的5個外顯子的上、下游引物,聚合酶鏈反應(PCR)擴增目標DNA序列。25.0 μl反應體系中,2倍Taq PCR標準混合物12.5 μl,10 μmol/L正、反向引物各1.0 μl,100 ng/μl DNA模板2.0 μl,超純水9.5 μl。95℃預變性5 min;94℃變性40 s,55℃退火30 s,72℃延伸1 min,共40個循環,最后72 ℃延伸5 min。PCR產物經1%瓊脂糖凝膠電泳,溴化乙錠和(或)紫外線鑒定,證實PCR擴增陽性片段存在。
采用ABI 3730xl型測序儀熒光標記法對PCR產物進行正、反向DNA測序。測序由深圳華大基因研究院杭州分公司完成。測序結果與Genbank、千人基因組計劃中的野生型CRX參考序列(GenBank: KR709702.1)進行BLAST比對分析,初步確定突變位點。應用Primer 5.0軟件設計針對突變位點的等位基因特異性(AS)-PCR引物(正向引物:CCCACCTCCCTA TCAGGCT;反向引物:GACGGTGTTAGGGTTGAG),驗證突變。
以150名無血緣關系的健康個體為對照組。應用SIFT功能預測軟件分析所發現的CRX基因突變可能的生物學效應。
2 結果
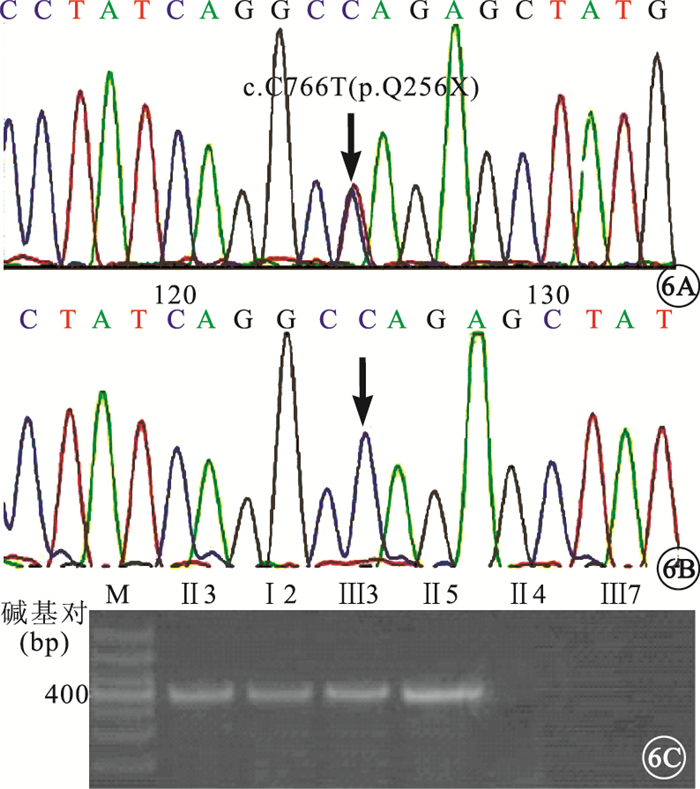
患者Ⅰ2、Ⅱ3、Ⅱ5、Ⅲ3的CRX第5外顯子均存在c.C766T (p.Q256X)無義突變(圖 6A),造成CRX蛋白第256位的谷氨酰胺突變為終止密碼子,形成截短的CRX蛋白。SIFT分析提示,該突變為有害性:damaging。
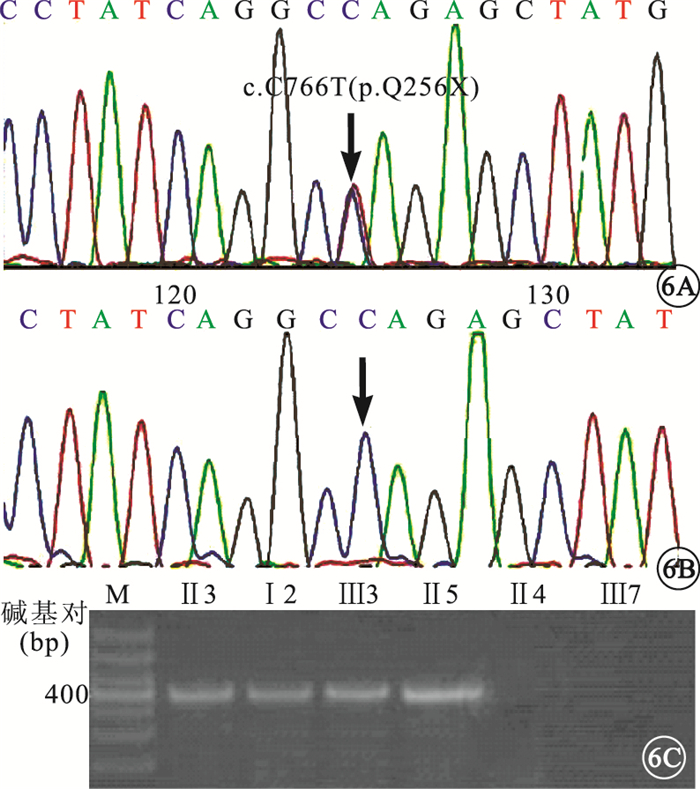 圖6
DNA測序圖和AS-PCR電泳圖。6A.患者CRX基因第5外顯子的c.C766T突變(黑箭);6B.對照組該位點不存在突變;6C.AS-PCR顯示,患者可擴增出406堿基對條帶,對照組無擴增條帶
圖6
DNA測序圖和AS-PCR電泳圖。6A.患者CRX基因第5外顯子的c.C766T突變(黑箭);6B.對照組該位點不存在突變;6C.AS-PCR顯示,患者可擴增出406堿基對條帶,對照組無擴增條帶
正常家系成員以及對照組均未見此基因突變(圖 6B)。AS-PCR驗證結果顯示,家系中基因突變攜帶者存在此突變位點;家系中正常成員和對照組中隨機個體均未見此突變位點(圖 6C)。
3 討論
CRX基因定位于19q13.32,包含5個外顯子,編碼含299個氨基酸殘基的CRX蛋白。CRX蛋白為光感受器細胞特異性的反式作用轉錄因子,調控早期光感受器細胞的發育和成熟光感受器細胞的存活[4-7]。CRX功能的喪失可影響視網膜內多種蛋白的表達,如光轉換通路相關蛋白、視蛋白、感光細胞結構蛋白和磷酸二酯酶等[4-7]。CRX突變可導致一系列的視網膜光感受器細胞遺傳性疾病,除CORD2外,還有視網膜色素變性和Leber先天性黑矇[8-11]。此3種疾病在眼科臨床上有不同的定義,但都可以由CRX基因突變導致,因而有理由認為3種疾病具有臨床異質性特點,可能具有相同的分子病因,只是在疾病嚴重度上和對視桿或視錐細胞的影響不同。
本研究本研究在對所有家系成員進行CRX基因突變的檢測中,發現4例患者的共有無義突變c.C766T (p.Gln256X),不僅為家系的產前DNA診斷、植入前遺傳學診斷等疾病預防措施奠定了基礎,而且補充和豐富了CRX基因突變數據庫。隨著分子遺傳學技術的進步, 收集典型家系尋找新的致病位點, 對CORD發病機制的探討以及分析基因型-表型相關性以預測疾病的發生和發展,將是今后研究的重點[2, 3, 5, 11]。
視錐-視桿營養不良(CORD)為高度臨床異質性和遺傳異質性眼病,以視錐細胞受損為主,伴不同程度的視桿細胞損傷[1-3]。目前, 已明確的CORD致病基因已有25個[1-3]。其中,常染色體隱性遺傳基因13個;常染色體顯性遺傳基因10個;X連鎖遺傳基因2個。本研究對1個南方漢族常染色體顯性遺傳CORD家系的視錐-視桿同源盒(CRX)基因突變譜進行了測序驗證。現將結果報道如下。
1 對象和方法
本研究經本院醫學倫理委員會審核批準。參與研究的所有個體均簽署知情同意書。
來自南方的1個4代19人漢族家系,其中CODR患者4例(圖 1)。對所有家系成員均行眼科檢查。先證者,女,52歲。10余年前雙眼開始視力進行性下降。右眼視力0.1,左眼視力0.15,均不能矯正。畏光但無眼球震顫;色覺輕微異常。眼底黃斑區2個視盤直徑(DD)×2 DD金箔樣反光,散在色素沉著,中心凹反光消失,伴黃色斑點;中周部視網膜可見骨細胞樣色素沉著,視網膜動脈血管變細(圖 2)。光相干斷層掃描(OCT)檢查可見黃斑區外層視網膜萎縮變薄(圖 3)。閃光視網膜電圖(FERG)檢查,暗適應b波振幅下降,視桿反應輕度下降;明適應a、b波振幅均明顯下降,視錐反應消失(圖 4)。熒光素眼底血管造影(FFA)檢查黃斑區呈強熒光,未見脈絡膜湮沒(圖 5)。家系其余3例患者中,男性1例,女性2例。年齡分別為75、48、32歲;發病年齡分別為40、39、32歲。視力0.05~0.4。均有畏光但無眼球震顫。色覺檢查,輕微異常、色盲、色弱各1例。眼底黃斑區1.0 DD×1.5 DD金箔樣反光,散在色素沉著1例;黃斑區色素沉著2例。中心凹反光消失,伴黃色斑點,視網膜色素沉著1例;中心凹反光不清2例,其中外周視網膜可見針尖樣骨細胞色素沉著1例。FERG檢查,視錐反應消失1例,明顯下降2例;視桿反應輕度下降2例,正常1例。其余家系成員檢查結果未見異常。依據臨床檢查結果和家系分析,結合國內外文獻[1-4],初步診斷為常染色體顯性遺傳CORD (CORD2)。
圖1
患者家系圖。■:男性患者;●:女性患者;↑:先證者;□:正常男性;○:正常女性
圖2
先證者右眼彩色眼底像。黃斑區不規則圓形黃色萎縮灶,暴露脈絡膜大血管,其間雜色素沉著,后極部見黃色斑點??圖 3??先證者右眼OCT像。黃斑區外層視網膜萎縮變薄??圖 4??先證者雙眼FERG像。4A.暗適應b波振幅下降,視桿反應輕度下降;4B.明適應a、b波振幅明顯下降,視錐反應消失
圖5
先證者雙眼FFA像。5A.左眼,早期黃斑區見強熒光,晚期呈透見熒光;5B.右眼,黃斑“靶的”狀強熒光,透見與遮蔽熒光相間,晚期色素上皮病變部位呈斑駁狀強熒光
采集家系所有成員的外周血5 ml,乙二胺四乙酸抗凝。天根血液基因組DNA抽提試劑盒(北京天根生化科技有限公司)提取基因組DNA。應用Primer 5.0軟件設計CRX基因的5個外顯子的上、下游引物,聚合酶鏈反應(PCR)擴增目標DNA序列。25.0 μl反應體系中,2倍Taq PCR標準混合物12.5 μl,10 μmol/L正、反向引物各1.0 μl,100 ng/μl DNA模板2.0 μl,超純水9.5 μl。95℃預變性5 min;94℃變性40 s,55℃退火30 s,72℃延伸1 min,共40個循環,最后72 ℃延伸5 min。PCR產物經1%瓊脂糖凝膠電泳,溴化乙錠和(或)紫外線鑒定,證實PCR擴增陽性片段存在。
采用ABI 3730xl型測序儀熒光標記法對PCR產物進行正、反向DNA測序。測序由深圳華大基因研究院杭州分公司完成。測序結果與Genbank、千人基因組計劃中的野生型CRX參考序列(GenBank: KR709702.1)進行BLAST比對分析,初步確定突變位點。應用Primer 5.0軟件設計針對突變位點的等位基因特異性(AS)-PCR引物(正向引物:CCCACCTCCCTA TCAGGCT;反向引物:GACGGTGTTAGGGTTGAG),驗證突變。
以150名無血緣關系的健康個體為對照組。應用SIFT功能預測軟件分析所發現的CRX基因突變可能的生物學效應。
2 結果
患者Ⅰ2、Ⅱ3、Ⅱ5、Ⅲ3的CRX第5外顯子均存在c.C766T (p.Q256X)無義突變(圖 6A),造成CRX蛋白第256位的谷氨酰胺突變為終止密碼子,形成截短的CRX蛋白。SIFT分析提示,該突變為有害性:damaging。
圖6
DNA測序圖和AS-PCR電泳圖。6A.患者CRX基因第5外顯子的c.C766T突變(黑箭);6B.對照組該位點不存在突變;6C.AS-PCR顯示,患者可擴增出406堿基對條帶,對照組無擴增條帶
正常家系成員以及對照組均未見此基因突變(圖 6B)。AS-PCR驗證結果顯示,家系中基因突變攜帶者存在此突變位點;家系中正常成員和對照組中隨機個體均未見此突變位點(圖 6C)。
3 討論
CRX基因定位于19q13.32,包含5個外顯子,編碼含299個氨基酸殘基的CRX蛋白。CRX蛋白為光感受器細胞特異性的反式作用轉錄因子,調控早期光感受器細胞的發育和成熟光感受器細胞的存活[4-7]。CRX功能的喪失可影響視網膜內多種蛋白的表達,如光轉換通路相關蛋白、視蛋白、感光細胞結構蛋白和磷酸二酯酶等[4-7]。CRX突變可導致一系列的視網膜光感受器細胞遺傳性疾病,除CORD2外,還有視網膜色素變性和Leber先天性黑矇[8-11]。此3種疾病在眼科臨床上有不同的定義,但都可以由CRX基因突變導致,因而有理由認為3種疾病具有臨床異質性特點,可能具有相同的分子病因,只是在疾病嚴重度上和對視桿或視錐細胞的影響不同。
本研究本研究在對所有家系成員進行CRX基因突變的檢測中,發現4例患者的共有無義突變c.C766T (p.Gln256X),不僅為家系的產前DNA診斷、植入前遺傳學診斷等疾病預防措施奠定了基礎,而且補充和豐富了CRX基因突變數據庫。隨著分子遺傳學技術的進步, 收集典型家系尋找新的致病位點, 對CORD發病機制的探討以及分析基因型-表型相關性以預測疾病的發生和發展,將是今后研究的重點[2, 3, 5, 11]。