引用本文: 郭慧青, 盛迅倫. 散發型視網膜色素變性致病基因及其遺傳方式觀察. 中華眼底病雜志, 2016, 32(6): 576-581. doi: 10.3760/cma.j.issn.1005-1015.2016.06.004 復制
視網膜色素變性(RP)遺傳方式復雜并且具有高度的遺傳異質性。除了常染色體顯性遺傳(AD)、常染色體隱性遺傳(AR)、X連鎖遺傳(XL)以及少數雙基因型及線粒體遺傳RP,還有40%~50%的RP患者因缺乏家族史而無遺傳規律可循,稱為散發型RP (sRP)[1, 2]。目前已證實,與RP有關的基因已超過70個[2],致病基因眾多且缺乏突變熱點。既往采用篩選單個候選基因的方法篩查sRP患者的致病基因及其突變位點,不僅耗時漫長且檢測效率低下,難以篩查到致病基因。目標序列捕獲測序是將目標序列捕獲芯片與高通量二代測序(NGS)技術相整合進行測序,以獲得目標基因組序列的研究策略,使得致病基因突變檢測更為簡便、高效和準確。本研究采用外顯子捕獲結合NGS技術對一組sRP患者進行了致病基因突變檢測,分析其遺傳方式。現將結果報道如下。
1 對象和方法
2013年3月至2014年10月在寧夏眼科醫院確診為sRP的患者49例以及家庭成員128名納入研究。本研究經本院倫理委員會審核批準。遵循赫爾辛基宣言,所有受檢者和未成年患者監護人均簽署知情同意書。
參照文獻[3, 4]確立本組患者納入標準,典型RP:(1)?有夜盲史;(2)?視力逐漸下降;(3)?典型眼底改變,視盤呈蠟黃色萎縮,視網膜有骨細胞樣色素沉著,血管變細,視網膜呈青灰色;(4)?早期周邊視野呈環形暗點,晚期視野呈向中心縮窄;(5)?病變早期即可出現視網膜電圖(ERG)、眼電圖(EOG)明顯異常。無色素性RP:有夜盲史,ERG、EOG異常;眼底無色素沉著或僅在周邊眼底出現少數幾個骨細胞樣色素斑。結晶樣RP (BCD):符合典型RP臨床表現,眼底改變表現為閃亮的黃白色結晶,伴色素沉著。Usher綜合征2型(USH2):符合典型RP眼底改變,且伴中等程度的耳聾,前庭功能正常[5, 6]。排除Bardet-Biedl綜合征和代謝異常所致的視網膜色素上皮(RPE)變性。
詳細收集患者病史和家族史;詢問病史確定夜盲出現年齡。所有患者及其家庭成員均行最佳矯正視力(BCVA)、裂隙燈顯微鏡、間接檢眼鏡、眼壓、眼底彩色照相、視野、光相干斷層掃描(OCT)、全視野ERG檢查。行眼底自身熒光和熒光素眼底血管造影檢查10例。所有患者病史、家族史及臨床表現符合sRP診斷標準[7, 8]。其家庭成員均無夜盲病史,眼底檢查未見色素沉著。
抽取患者及其家庭成員外周靜脈血3~5 ml, 乙二胺四乙酸抗凝,采用德國QIAGEN公司Qiamp Blood試劑盒按照操作規程提取全基因組DNA。DNA質量濃度≥50 ng/μl, 吸光度[A,舊稱光密度(OD)]A260/A280為1.8~2.0,DNA總量≥6 μg。應用美國Agilent公司Sureselect RNA探針對目前已知的230個遺傳性視網膜疾病(HRD)致病基因的外顯子區域進行捕獲。針對HRD 2560個外顯子非重復區設計生物素化的單鏈捕獲探針[9]。將1 μg DNA文庫與緩沖BL液和設計的探針混合,95℃加熱7 min,65℃加熱2 min; 加入23 ml預熱至65℃的HY緩沖液,65℃雜交22 h。用500 μl 1倍結合緩沖液清洗50 μl MyOne磁珠(美國Life Technology公司) 3次,重懸于80 μl 1倍結合緩沖液。加入64 μl 2倍結合緩沖液至雜交混合物中,轉移至含有80 μl MyOne磁珠的試管中。旋轉混勻1 h。WB1緩沖液室溫清洗磁珠15 min,WB3緩沖液65℃清洗3次,15 min/次。洗脫緩沖液洗脫結合的DNA。洗脫的DNA進行聚合酶鏈反應(PCR),98℃預變性30 s; 98℃變性25 s,65℃退火30 s,72℃延伸30 s,共進行15個循環;最后72℃延伸5 min。純化PCR產物。富集的文庫用Il-lumina HiSeq 2000第二代測序儀進行雙端測序,讀長為100堿基對。
目標序列測序后原始數據經過拆分獲得各個樣本的原始序列,去除測序數據中的接頭和質量值≤20的低質量數據,運用BWA軟件對比參考基因,應用GATK軟件對各個樣本的比對數據進行多態性位點檢測,對單個堿基的變異和單核苷酸多態性(SNPs)插入缺失突變(InDels)等數據進行統計分析。同時對測序深度、均一性、探針特異性等數據進行統計和分析。去除dbSNP137數據庫、千人基因組和內部數據庫800名正常漢族人數據庫中出現的SNPs和InDels。同時過濾掉SIFT預測對蛋白功能無影響的突變位點作為最后疾病相關的突變位點。根據AD、AR、XL的遺傳規律,分析家族史,確立其遺傳類型[10, 11]。
根據需要測序的DNA片段合成引物,PCR進行擴增,美國Applied Biosystems公司ABI3730xl測序儀以Sanger測序法進行測序,并將測序結果與目標序列捕獲測序后的結果進行比對,最終確定致病基因突變位點。
2 結果
設計合成的目標基因特異性捕獲探針可有效捕捉并富集基因組DNA的目標靶片段。49例患者目標序列平均測序深度為52.93~57.15,目標序列覆蓋率71.0%~140.0%。98.17%~99.08%的目標序列覆蓋率 > 4×,96.79%~98.48%目標序列覆蓋率 > 10×,92.42%~96.90%的目標序列覆蓋率 > 20×。
49例患者中,24例患者成功檢測到致病突變位點,共涉及16個基因;檢測陽性率為49.0%。發現突變位點41個。其中,新發現突變位點32個,占78.0%(表 1)。
24例患者中,致病基因USH2A 7例,占29.2%;C2orf71、RDH12、GNGA1各2例,分別占8.3%RPGR1、IFT140、TULP1、CLRN1、RPE65、ABCA4、GUCA1A、EYS、CYP4V2、GPR98、ATXN7各1例,分別占4.2%。根據其家系成員基因篩查結果分析,24例患者中,ARRP者20例,占83.3%;ADRP者3例,占12.5%;XLRP者1例,占4.2%。7例檢測到USH2A基因突變的患者中,確診為USH2者3例,占所有患者的6.1%;無綜合征ARRP者4例,占所有患者的8.2%。
24例患者出現夜盲年齡為2~47歲,就診時年齡為4~70歲。BCVA為眼前數指~1.0。
ARRP者20例,患者序號為2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、20、22、23。致病基因分別為USH2A、IFT140、USH2A、C2orf71、CNGA1、C2orf71、RDH12、USH2A、TULP1、CLRN1、CNGA1、RPE65、USH2A、USH2A、USH2A、USH2A、ABCA4、EYS、CYP4V2、GPR98。出現夜盲
時年齡5~40歲。BCVA為眼前數指~1.0。眼底視盤顏色明顯變淡或蠟黃,視網膜血管變細,后極部及周邊散在骨細胞樣色素沉著;視網膜神經上皮層變薄。黃斑部萎縮7例,伴黃斑前膜2例,伴黃斑囊樣水腫1例。全視野ERG檢查,暗適應a、b波振幅中度、重度下降1、6例,熄滅狀12例;明適應a、b波振幅中度、重度下降3、7例,熄滅狀9例。未行全視野ERG檢查1例。色覺檢查:全色盲、紅綠色盲各1例;色覺正常11例;未行色覺檢查7例。
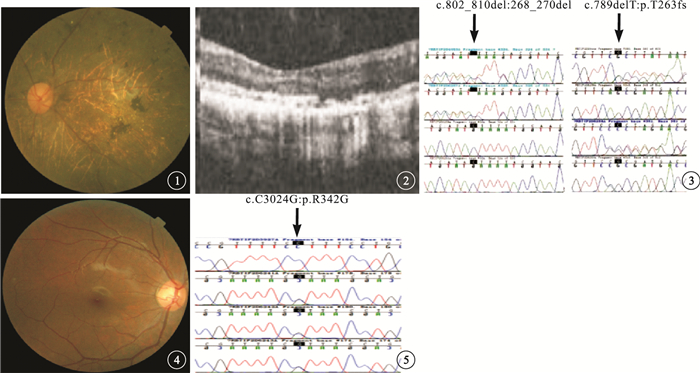
序號22患者,眼底視盤顏色蠟黃;視網膜血管變細,后極部可見結晶樣亮點,后極部包括黃斑區散在分布結晶樣亮點或融合成條狀,大小約為視網膜中央靜脈管徑的一半,視網膜散在不規則樣色素沉著(圖 1)。OCT檢查可見中心凹形態變平,神經上皮層變薄,橢圓體帶缺失,RPE層不連續(圖 2)。臨床診斷為BCD。基因測序為CYP4V2基因的復合雜合性突變(p.268-270del,c.789delT)(圖 3),為新發現的突變位點。
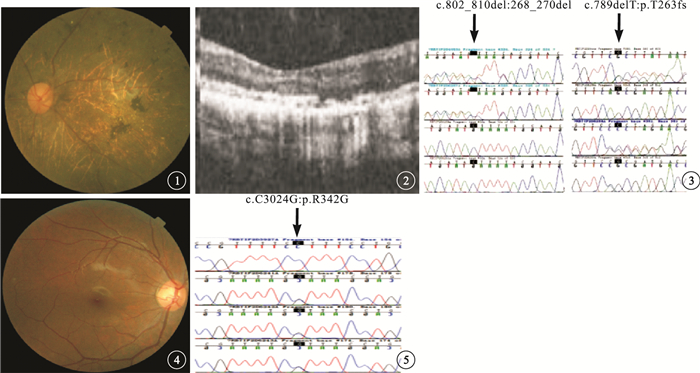 圖1
左眼彩色眼底像。視盤顏色蠟黃,視網膜血管明顯變細,后極部散在分布結晶樣亮點,以及散在不規則樣色素沉著??圖 2?左眼OCT像。黃斑中心凹形態變平,神經上皮層變薄,橢圓體帶缺失,RPE層不連續??圖 3?基因測序圖。CYP4V2基因復合雜合性突變(p.268-270del,c.789delT)(黑箭)??圖 4?右眼彩色眼底像。視盤顏色紅潤,視網膜內未見色素沉著??圖 5?基因測序圖。TULP1基因的純合突變(c.C1024G)(黑箭)
圖1
左眼彩色眼底像。視盤顏色蠟黃,視網膜血管明顯變細,后極部散在分布結晶樣亮點,以及散在不規則樣色素沉著??圖 2?左眼OCT像。黃斑中心凹形態變平,神經上皮層變薄,橢圓體帶缺失,RPE層不連續??圖 3?基因測序圖。CYP4V2基因復合雜合性突變(p.268-270del,c.789delT)(黑箭)??圖 4?右眼彩色眼底像。視盤顏色紅潤,視網膜內未見色素沉著??圖 5?基因測序圖。TULP1基因的純合突變(c.C1024G)(黑箭)
序號10患兒,5歲。夜間視力差。眼底視盤顏色紅潤,視網膜未見明顯色素沉著(圖 4)。OCT、眼底自身熒光檢查均未見異常。臨床診斷為無色素性RP。基因測序為TULP1基因的純合突變(c.C1024G)(圖 5)。其父母各攜帶1條突變基因;其姐姐未訴夜間視力差。眼底未見明顯色素沉著。ERG檢查雙眼b波中度下降,其余未見異常。基因測序結果為攜帶1條TULP1基因的錯義突變(c.C1024G)。
ADRP者3例,患者序號為19、21、24。致病基因分別為GUCA1A、RDH12、ATXN4;突變位點分別為c.T466C、c.C532T、c.G746T,其中c.G746T為新發現錯義突變。患者出現夜盲時年齡分別為43、47、40歲。BCVA為眼前數指~1.0。眼底視盤顏色明顯變淡或蠟黃,視網膜血管變細,后極部及周邊散在骨細胞樣色素顆粒沉著。全視野ERG檢查,暗適應a、b波振幅中、重度下降各1例;明適應a、b波振幅中度下降、熄滅狀各1例。未行全視野ERG檢查1例。
XLRP者1例,患者序號1。致病基因RPGR;突變位點c.2405_2406del,為新發現移碼突變。患者出現夜盲時年齡20歲。BCVA為0.25。眼底視盤顏色淡,視網膜血管變細,周邊彌漫性骨細胞樣色素顆粒沉著。OCT檢查顯示神經上皮層變薄。色覺檢查正常。全視野ERG檢查,暗適應、明適應a、b波振幅均呈熄滅狀。
致病基因USH2A 7例中,確診為USH2綜合征者3例,患者序號為2、4、15。致病基因USH2A。其中,序號2患者該基因的突變類型為2個錯義突變和1個剪切突變;序號4患者該基因的突變類型為2個錯義突變和1個移碼突變;序號15患者該基因的突變類型為復合雜合型錯義突變。患者均自幼出現中等程度聽力下降,前庭功能檢查均正常。眼底視盤顏色淡,視網膜血管變細,周邊骨細胞樣色素沉著。OCT檢查顯示神經上皮層變薄。全視野ERG檢查,暗適應a、b波振幅中、重度下降、熄滅狀各1例;明適應a、b波振幅中度下降、重度下降、熄滅狀各1例。
3 討論
RP致病基因與臨床表型具有高度異質性。應用連鎖分析等傳統方法研究孟德爾遺傳方式的遺傳性疾病,一般需要足夠多的患者或大家系,對于小家系或散發患者難以分析。近年出現的外顯子捕獲技術利用特制的探針對特定的蛋白編碼區域DNA或某段特定的序列進行捕獲,富集后再進行高通量測序,具有效率高、經費低、時間短等優點[9]。我們前期曾使用傳統測序方法對87例sRP患者檢測了常見的5個RP致病突變基因,未發現致病突變基因位點[12]。本研究采用外顯子結合目標區域測序技術,49例患者中24例患者成功檢測到致病突變基因位點,共涉及16個基因,檢測陽性率為49.0%,極大提高檢測的效率。但仍有51%的患者未找到基因突變位點,其原因一方面可能是檢測方法仍存在一定局限性;另一方面也不能排除此部分患者中存在新的RP致病基因的可能。
Hartong等[10]認為,多數sRP患者為AR或XL (對于男性患者)。盡管少數患者可能是由于自身基因突變而開始一個新的遺傳過程,即由該基因突變患者作為初始進行遺傳,在以后的遺傳中,遺傳方式可以為ADRP、ARRP、XLRP遺傳類型中的任何一種。本研究檢測到致病基因的24例患者中,83.3%為AR,12.5%為AD,4.2%為XL。與Hartong等[10]觀點相符。
本研究中24例患者檢測到致病突變基因16個,突變位點41個,其中78.0%為新發現的突變位點。由于種族、地域不同,不同種族人群的突變頻譜存在較大差異[12]。有學者報道,日本人群sRP和ARRP中GNGA1、EYS是主要致病基因,突變率分別為13.3%、13.3%;GNGA1的突變率明顯高于歐洲國家[13]。歐洲人群EYS、USH2A在sRP、ARRP患者中突變率較高[14]。本研究中涉及的突變基因較多,其中USH2A基因的突變率較高,占29.2%,其他各基因突變率較低。因此,我們認為USH2A基因是本組sRP患者的主要致病基因。但由于樣本量不夠大,還需進一步研究結果證實。
RP可以合并不同類型的綜合征,其中最常見者為USH;大約可占全部RP患者的15.0%~20.0%[5]。根據臨床表現不同USH又可分為3型。USH2A基因是USH2的主要致病基因,約占USH2致病基因的70.0%。目前已發現100余種突變與之相關[6]。目前國內外已有多項研究表明,USH2A基因突變也可引起非綜合征型的常染色體ARRP,且較為常見[15]。Seyedahmadi等[16]對北美225例非綜合征型ARRP患者進行USH2A基因突變分析,結果發現有4.5%的患者攜帶錯義突變p.C759F,且與USH2患者的突變類型有所不同。非綜合征RP患者USH2A基因突變形式通常為純合錯義突變或復合雜合錯義突變。美國有研究者對USH和非綜合征ARRP患者進行基因突變分析,發現12.0%~25.0%的ARRP患者攜帶USH2A基因突變[17]。由此可見,USH2A基因是美國ARRP患者中最常見的致病基因。本研究中,共有7例患者檢測到USH2A基因突變,3例患者自幼出現中等程度的聽力損害,前庭功能正常診斷為USH2。其中序號2患者該基因的突變類型為2個錯義突變和1個剪切突變,序號4患者該基因的突變類型為2個錯義突變和1個移碼突變;序號15患者為復合雜合型錯義突變。另外4例患者無聽力異常,診斷為無綜合征型ARRP。突變方式均為復合雜合型錯義突變。目前,在國內關于USH2A基因的研究較少,且無有關USH2A基因可引起無綜合征型ARRP的報道。本研究中,USH2A基因在無綜合征型ARRP患者中的突變率較高,占8.2%。
RP分為典型性和非典型性。非典型性RP包括BCD、無色素性RP、中心性和旁中心性RP和單眼RP[16, 17]。本研究中BCD和無色素性RP各1例。BCD在歐美國家發病率較低,而在中國和日本發病率則相對較高,中國人群中其發病率為1/24 000[18]。目前,唯一可確定的導致BCD的基因是CYP4V2,其編碼產物參與脂肪酸的代謝[18, 19]。單明華和李揚[19]對國內24例BCD患者進行CYP4V2基因檢測,除1例患者外,其他患者均檢測到CYP4V2基因上的突變。本研究中序號22患者臨床診斷為BCD,基因測序結果為CYP4V2基因上的2個移碼突變(c.802-810del、c.789delT),為未報道過的新發突變。進一步證實了CYP4V2基因突變可導致BCD,擴展了CYP4V2的基因突變譜。無色素性RP患者臨床特征為夜盲但眼底無色素沉著,也有部分患者長期觀察可出現色素沉著。本研究中序號10患兒為無色素性RP,基因測序結果為TULP1基因上的純合錯義突變(c.C1024G)。其父母各攜帶1條突變基因。其姐姐未訴夜間視力差,眼底未見明顯色素沉著,ERG檢查b波雙眼中度下降。基因測序結果為攜帶者。目前國內有關無色素性RP基因突變研究較少。有研究報道,TULP1基因突變是與早發型RP有關的1個基因[20],但國內暫無TULP1基因突變與無色素性RP的相關研究報道。
視網膜色素變性(RP)遺傳方式復雜并且具有高度的遺傳異質性。除了常染色體顯性遺傳(AD)、常染色體隱性遺傳(AR)、X連鎖遺傳(XL)以及少數雙基因型及線粒體遺傳RP,還有40%~50%的RP患者因缺乏家族史而無遺傳規律可循,稱為散發型RP (sRP)[1, 2]。目前已證實,與RP有關的基因已超過70個[2],致病基因眾多且缺乏突變熱點。既往采用篩選單個候選基因的方法篩查sRP患者的致病基因及其突變位點,不僅耗時漫長且檢測效率低下,難以篩查到致病基因。目標序列捕獲測序是將目標序列捕獲芯片與高通量二代測序(NGS)技術相整合進行測序,以獲得目標基因組序列的研究策略,使得致病基因突變檢測更為簡便、高效和準確。本研究采用外顯子捕獲結合NGS技術對一組sRP患者進行了致病基因突變檢測,分析其遺傳方式。現將結果報道如下。
1 對象和方法
2013年3月至2014年10月在寧夏眼科醫院確診為sRP的患者49例以及家庭成員128名納入研究。本研究經本院倫理委員會審核批準。遵循赫爾辛基宣言,所有受檢者和未成年患者監護人均簽署知情同意書。
參照文獻[3, 4]確立本組患者納入標準,典型RP:(1)?有夜盲史;(2)?視力逐漸下降;(3)?典型眼底改變,視盤呈蠟黃色萎縮,視網膜有骨細胞樣色素沉著,血管變細,視網膜呈青灰色;(4)?早期周邊視野呈環形暗點,晚期視野呈向中心縮窄;(5)?病變早期即可出現視網膜電圖(ERG)、眼電圖(EOG)明顯異常。無色素性RP:有夜盲史,ERG、EOG異常;眼底無色素沉著或僅在周邊眼底出現少數幾個骨細胞樣色素斑。結晶樣RP (BCD):符合典型RP臨床表現,眼底改變表現為閃亮的黃白色結晶,伴色素沉著。Usher綜合征2型(USH2):符合典型RP眼底改變,且伴中等程度的耳聾,前庭功能正常[5, 6]。排除Bardet-Biedl綜合征和代謝異常所致的視網膜色素上皮(RPE)變性。
詳細收集患者病史和家族史;詢問病史確定夜盲出現年齡。所有患者及其家庭成員均行最佳矯正視力(BCVA)、裂隙燈顯微鏡、間接檢眼鏡、眼壓、眼底彩色照相、視野、光相干斷層掃描(OCT)、全視野ERG檢查。行眼底自身熒光和熒光素眼底血管造影檢查10例。所有患者病史、家族史及臨床表現符合sRP診斷標準[7, 8]。其家庭成員均無夜盲病史,眼底檢查未見色素沉著。
抽取患者及其家庭成員外周靜脈血3~5 ml, 乙二胺四乙酸抗凝,采用德國QIAGEN公司Qiamp Blood試劑盒按照操作規程提取全基因組DNA。DNA質量濃度≥50 ng/μl, 吸光度[A,舊稱光密度(OD)]A260/A280為1.8~2.0,DNA總量≥6 μg。應用美國Agilent公司Sureselect RNA探針對目前已知的230個遺傳性視網膜疾病(HRD)致病基因的外顯子區域進行捕獲。針對HRD 2560個外顯子非重復區設計生物素化的單鏈捕獲探針[9]。將1 μg DNA文庫與緩沖BL液和設計的探針混合,95℃加熱7 min,65℃加熱2 min; 加入23 ml預熱至65℃的HY緩沖液,65℃雜交22 h。用500 μl 1倍結合緩沖液清洗50 μl MyOne磁珠(美國Life Technology公司) 3次,重懸于80 μl 1倍結合緩沖液。加入64 μl 2倍結合緩沖液至雜交混合物中,轉移至含有80 μl MyOne磁珠的試管中。旋轉混勻1 h。WB1緩沖液室溫清洗磁珠15 min,WB3緩沖液65℃清洗3次,15 min/次。洗脫緩沖液洗脫結合的DNA。洗脫的DNA進行聚合酶鏈反應(PCR),98℃預變性30 s; 98℃變性25 s,65℃退火30 s,72℃延伸30 s,共進行15個循環;最后72℃延伸5 min。純化PCR產物。富集的文庫用Il-lumina HiSeq 2000第二代測序儀進行雙端測序,讀長為100堿基對。
目標序列測序后原始數據經過拆分獲得各個樣本的原始序列,去除測序數據中的接頭和質量值≤20的低質量數據,運用BWA軟件對比參考基因,應用GATK軟件對各個樣本的比對數據進行多態性位點檢測,對單個堿基的變異和單核苷酸多態性(SNPs)插入缺失突變(InDels)等數據進行統計分析。同時對測序深度、均一性、探針特異性等數據進行統計和分析。去除dbSNP137數據庫、千人基因組和內部數據庫800名正常漢族人數據庫中出現的SNPs和InDels。同時過濾掉SIFT預測對蛋白功能無影響的突變位點作為最后疾病相關的突變位點。根據AD、AR、XL的遺傳規律,分析家族史,確立其遺傳類型[10, 11]。
根據需要測序的DNA片段合成引物,PCR進行擴增,美國Applied Biosystems公司ABI3730xl測序儀以Sanger測序法進行測序,并將測序結果與目標序列捕獲測序后的結果進行比對,最終確定致病基因突變位點。
2 結果
設計合成的目標基因特異性捕獲探針可有效捕捉并富集基因組DNA的目標靶片段。49例患者目標序列平均測序深度為52.93~57.15,目標序列覆蓋率71.0%~140.0%。98.17%~99.08%的目標序列覆蓋率 > 4×,96.79%~98.48%目標序列覆蓋率 > 10×,92.42%~96.90%的目標序列覆蓋率 > 20×。
49例患者中,24例患者成功檢測到致病突變位點,共涉及16個基因;檢測陽性率為49.0%。發現突變位點41個。其中,新發現突變位點32個,占78.0%(表 1)。
24例患者中,致病基因USH2A 7例,占29.2%;C2orf71、RDH12、GNGA1各2例,分別占8.3%RPGR1、IFT140、TULP1、CLRN1、RPE65、ABCA4、GUCA1A、EYS、CYP4V2、GPR98、ATXN7各1例,分別占4.2%。根據其家系成員基因篩查結果分析,24例患者中,ARRP者20例,占83.3%;ADRP者3例,占12.5%;XLRP者1例,占4.2%。7例檢測到USH2A基因突變的患者中,確診為USH2者3例,占所有患者的6.1%;無綜合征ARRP者4例,占所有患者的8.2%。
24例患者出現夜盲年齡為2~47歲,就診時年齡為4~70歲。BCVA為眼前數指~1.0。
ARRP者20例,患者序號為2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、20、22、23。致病基因分別為USH2A、IFT140、USH2A、C2orf71、CNGA1、C2orf71、RDH12、USH2A、TULP1、CLRN1、CNGA1、RPE65、USH2A、USH2A、USH2A、USH2A、ABCA4、EYS、CYP4V2、GPR98。出現夜盲
時年齡5~40歲。BCVA為眼前數指~1.0。眼底視盤顏色明顯變淡或蠟黃,視網膜血管變細,后極部及周邊散在骨細胞樣色素沉著;視網膜神經上皮層變薄。黃斑部萎縮7例,伴黃斑前膜2例,伴黃斑囊樣水腫1例。全視野ERG檢查,暗適應a、b波振幅中度、重度下降1、6例,熄滅狀12例;明適應a、b波振幅中度、重度下降3、7例,熄滅狀9例。未行全視野ERG檢查1例。色覺檢查:全色盲、紅綠色盲各1例;色覺正常11例;未行色覺檢查7例。
序號22患者,眼底視盤顏色蠟黃;視網膜血管變細,后極部可見結晶樣亮點,后極部包括黃斑區散在分布結晶樣亮點或融合成條狀,大小約為視網膜中央靜脈管徑的一半,視網膜散在不規則樣色素沉著(圖 1)。OCT檢查可見中心凹形態變平,神經上皮層變薄,橢圓體帶缺失,RPE層不連續(圖 2)。臨床診斷為BCD。基因測序為CYP4V2基因的復合雜合性突變(p.268-270del,c.789delT)(圖 3),為新發現的突變位點。
圖1
左眼彩色眼底像。視盤顏色蠟黃,視網膜血管明顯變細,后極部散在分布結晶樣亮點,以及散在不規則樣色素沉著??圖 2?左眼OCT像。黃斑中心凹形態變平,神經上皮層變薄,橢圓體帶缺失,RPE層不連續??圖 3?基因測序圖。CYP4V2基因復合雜合性突變(p.268-270del,c.789delT)(黑箭)??圖 4?右眼彩色眼底像。視盤顏色紅潤,視網膜內未見色素沉著??圖 5?基因測序圖。TULP1基因的純合突變(c.C1024G)(黑箭)
序號10患兒,5歲。夜間視力差。眼底視盤顏色紅潤,視網膜未見明顯色素沉著(圖 4)。OCT、眼底自身熒光檢查均未見異常。臨床診斷為無色素性RP。基因測序為TULP1基因的純合突變(c.C1024G)(圖 5)。其父母各攜帶1條突變基因;其姐姐未訴夜間視力差。眼底未見明顯色素沉著。ERG檢查雙眼b波中度下降,其余未見異常。基因測序結果為攜帶1條TULP1基因的錯義突變(c.C1024G)。
ADRP者3例,患者序號為19、21、24。致病基因分別為GUCA1A、RDH12、ATXN4;突變位點分別為c.T466C、c.C532T、c.G746T,其中c.G746T為新發現錯義突變。患者出現夜盲時年齡分別為43、47、40歲。BCVA為眼前數指~1.0。眼底視盤顏色明顯變淡或蠟黃,視網膜血管變細,后極部及周邊散在骨細胞樣色素顆粒沉著。全視野ERG檢查,暗適應a、b波振幅中、重度下降各1例;明適應a、b波振幅中度下降、熄滅狀各1例。未行全視野ERG檢查1例。
XLRP者1例,患者序號1。致病基因RPGR;突變位點c.2405_2406del,為新發現移碼突變。患者出現夜盲時年齡20歲。BCVA為0.25。眼底視盤顏色淡,視網膜血管變細,周邊彌漫性骨細胞樣色素顆粒沉著。OCT檢查顯示神經上皮層變薄。色覺檢查正常。全視野ERG檢查,暗適應、明適應a、b波振幅均呈熄滅狀。
致病基因USH2A 7例中,確診為USH2綜合征者3例,患者序號為2、4、15。致病基因USH2A。其中,序號2患者該基因的突變類型為2個錯義突變和1個剪切突變;序號4患者該基因的突變類型為2個錯義突變和1個移碼突變;序號15患者該基因的突變類型為復合雜合型錯義突變。患者均自幼出現中等程度聽力下降,前庭功能檢查均正常。眼底視盤顏色淡,視網膜血管變細,周邊骨細胞樣色素沉著。OCT檢查顯示神經上皮層變薄。全視野ERG檢查,暗適應a、b波振幅中、重度下降、熄滅狀各1例;明適應a、b波振幅中度下降、重度下降、熄滅狀各1例。
3 討論
RP致病基因與臨床表型具有高度異質性。應用連鎖分析等傳統方法研究孟德爾遺傳方式的遺傳性疾病,一般需要足夠多的患者或大家系,對于小家系或散發患者難以分析。近年出現的外顯子捕獲技術利用特制的探針對特定的蛋白編碼區域DNA或某段特定的序列進行捕獲,富集后再進行高通量測序,具有效率高、經費低、時間短等優點[9]。我們前期曾使用傳統測序方法對87例sRP患者檢測了常見的5個RP致病突變基因,未發現致病突變基因位點[12]。本研究采用外顯子結合目標區域測序技術,49例患者中24例患者成功檢測到致病突變基因位點,共涉及16個基因,檢測陽性率為49.0%,極大提高檢測的效率。但仍有51%的患者未找到基因突變位點,其原因一方面可能是檢測方法仍存在一定局限性;另一方面也不能排除此部分患者中存在新的RP致病基因的可能。
Hartong等[10]認為,多數sRP患者為AR或XL (對于男性患者)。盡管少數患者可能是由于自身基因突變而開始一個新的遺傳過程,即由該基因突變患者作為初始進行遺傳,在以后的遺傳中,遺傳方式可以為ADRP、ARRP、XLRP遺傳類型中的任何一種。本研究檢測到致病基因的24例患者中,83.3%為AR,12.5%為AD,4.2%為XL。與Hartong等[10]觀點相符。
本研究中24例患者檢測到致病突變基因16個,突變位點41個,其中78.0%為新發現的突變位點。由于種族、地域不同,不同種族人群的突變頻譜存在較大差異[12]。有學者報道,日本人群sRP和ARRP中GNGA1、EYS是主要致病基因,突變率分別為13.3%、13.3%;GNGA1的突變率明顯高于歐洲國家[13]。歐洲人群EYS、USH2A在sRP、ARRP患者中突變率較高[14]。本研究中涉及的突變基因較多,其中USH2A基因的突變率較高,占29.2%,其他各基因突變率較低。因此,我們認為USH2A基因是本組sRP患者的主要致病基因。但由于樣本量不夠大,還需進一步研究結果證實。
RP可以合并不同類型的綜合征,其中最常見者為USH;大約可占全部RP患者的15.0%~20.0%[5]。根據臨床表現不同USH又可分為3型。USH2A基因是USH2的主要致病基因,約占USH2致病基因的70.0%。目前已發現100余種突變與之相關[6]。目前國內外已有多項研究表明,USH2A基因突變也可引起非綜合征型的常染色體ARRP,且較為常見[15]。Seyedahmadi等[16]對北美225例非綜合征型ARRP患者進行USH2A基因突變分析,結果發現有4.5%的患者攜帶錯義突變p.C759F,且與USH2患者的突變類型有所不同。非綜合征RP患者USH2A基因突變形式通常為純合錯義突變或復合雜合錯義突變。美國有研究者對USH和非綜合征ARRP患者進行基因突變分析,發現12.0%~25.0%的ARRP患者攜帶USH2A基因突變[17]。由此可見,USH2A基因是美國ARRP患者中最常見的致病基因。本研究中,共有7例患者檢測到USH2A基因突變,3例患者自幼出現中等程度的聽力損害,前庭功能正常診斷為USH2。其中序號2患者該基因的突變類型為2個錯義突變和1個剪切突變,序號4患者該基因的突變類型為2個錯義突變和1個移碼突變;序號15患者為復合雜合型錯義突變。另外4例患者無聽力異常,診斷為無綜合征型ARRP。突變方式均為復合雜合型錯義突變。目前,在國內關于USH2A基因的研究較少,且無有關USH2A基因可引起無綜合征型ARRP的報道。本研究中,USH2A基因在無綜合征型ARRP患者中的突變率較高,占8.2%。
RP分為典型性和非典型性。非典型性RP包括BCD、無色素性RP、中心性和旁中心性RP和單眼RP[16, 17]。本研究中BCD和無色素性RP各1例。BCD在歐美國家發病率較低,而在中國和日本發病率則相對較高,中國人群中其發病率為1/24 000[18]。目前,唯一可確定的導致BCD的基因是CYP4V2,其編碼產物參與脂肪酸的代謝[18, 19]。單明華和李揚[19]對國內24例BCD患者進行CYP4V2基因檢測,除1例患者外,其他患者均檢測到CYP4V2基因上的突變。本研究中序號22患者臨床診斷為BCD,基因測序結果為CYP4V2基因上的2個移碼突變(c.802-810del、c.789delT),為未報道過的新發突變。進一步證實了CYP4V2基因突變可導致BCD,擴展了CYP4V2的基因突變譜。無色素性RP患者臨床特征為夜盲但眼底無色素沉著,也有部分患者長期觀察可出現色素沉著。本研究中序號10患兒為無色素性RP,基因測序結果為TULP1基因上的純合錯義突變(c.C1024G)。其父母各攜帶1條突變基因。其姐姐未訴夜間視力差,眼底未見明顯色素沉著,ERG檢查b波雙眼中度下降。基因測序結果為攜帶者。目前國內有關無色素性RP基因突變研究較少。有研究報道,TULP1基因突變是與早發型RP有關的1個基因[20],但國內暫無TULP1基因突變與無色素性RP的相關研究報道。