引用本文: 李憶安, 張琦, 彭婕, 黃秋婧, 趙培泉. 卷曲蛋白4聯合低密度脂蛋白相關蛋白5基因突變的家族性滲出性玻璃體視網膜病變二例. 中華眼底病雜志, 2016, 32(1): 79-81. doi: 10.3760/cma.j.issn.1005-1015.2016.01.019 復制
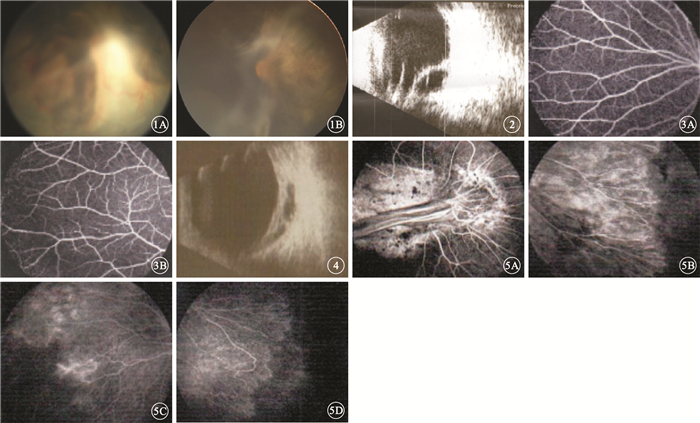
例1 患兒女,7個月。因發現雙眼斜視3個月就診。出生史G1P1,足月順產,出生體重2900 g;否認吸氧史。骨骼等全身檢查未見異常;新生兒聽力篩查正常。眼科檢查:雙眼眼前節正常。散瞳后應用第三代廣角數碼視網膜成像系統行眼底照相檢查,可見右眼視網膜皺襞連接至晶狀體后伴增生(
例2 患兒女,9歲。因左眼自幼視物不見就診。出生史G1P1,足月順產,出生體重3500 g;否認吸氧史。骨骼等全身檢查未見異常;聽力正常。眼科檢查:右眼視力0.02,左眼視力光感。雙眼眼壓均為13 mmHg(1 mmHg=0.133 kPa)。右眼眼前節正常;左眼晶狀體缺如,其余眼前節正常。眼底檢查,右眼視盤發出的鐮狀皺襞連接至晶狀體后;左眼全RD。眼B型超聲檢查,右眼顳下鐮狀RD伴機化膜;左眼全RD伴玻璃體積血或滲出,眼球壁鈣化(
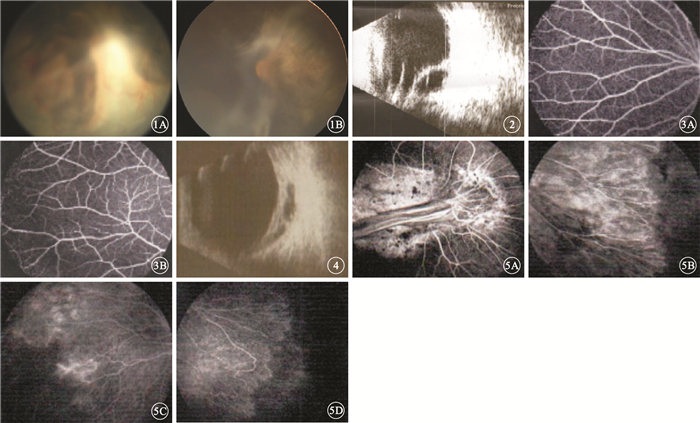 圖1
雙眼彩色眼底像。1A.右眼,視盤發出的皺襞連至晶狀體后;1B.左眼,視盤血管向顳側牽引伴纖維增生??圖 2?右眼B型超聲像。玻璃體內細帶狀回聲連接視盤至眼前節;玻璃體內機化增生膜與視網膜粘連牽拉,可見部分RD??圖 3?患兒父母FFA像。3A.父親左眼眼底周邊FFA像,血管分支較多,余未見明顯異常;3B.母親左眼周邊FFA像,血管分支較多,走形稍直,其余未見明顯異常??圖 4?右眼B型超聲像。顳下鐮狀RD伴機化膜??圖 5?患兒及父母FFA像。5A, 5B.患兒右眼FFA像,視盤邊界不清,形態異常,向顳側形成一鐮狀RD,其上可見血管;后極部動靜脈充盈尚可,血管僵直,近赤道分支眾多,呈扇形分布,部分血管熒光著染,周邊見近360°無血管區;5C, 5D.母親左右眼周邊FFA像,視網膜血管充盈尚可,血管分支增多,周邊顳側、顳上、顳下均見無血管區;左眼顳下血管末梢吻合,右眼顳下血管少量熒光著染
圖1
雙眼彩色眼底像。1A.右眼,視盤發出的皺襞連至晶狀體后;1B.左眼,視盤血管向顳側牽引伴纖維增生??圖 2?右眼B型超聲像。玻璃體內細帶狀回聲連接視盤至眼前節;玻璃體內機化增生膜與視網膜粘連牽拉,可見部分RD??圖 3?患兒父母FFA像。3A.父親左眼眼底周邊FFA像,血管分支較多,余未見明顯異常;3B.母親左眼周邊FFA像,血管分支較多,走形稍直,其余未見明顯異常??圖 4?右眼B型超聲像。顳下鐮狀RD伴機化膜??圖 5?患兒及父母FFA像。5A, 5B.患兒右眼FFA像,視盤邊界不清,形態異常,向顳側形成一鐮狀RD,其上可見血管;后極部動靜脈充盈尚可,血管僵直,近赤道分支眾多,呈扇形分布,部分血管熒光著染,周邊見近360°無血管區;5C, 5D.母親左右眼周邊FFA像,視網膜血管充盈尚可,血管分支增多,周邊顳側、顳上、顳下均見無血管區;左眼顳下血管末梢吻合,右眼顳下血管少量熒光著染
 圖6
基因突變序列圖。6A.FZD4FZD4基因;6B.LRP5基因。患兒c.1482發生G>A無義突變,為雜合子,LRP5基因c.3538發生G>A錯義突變,為雜合子;其父LRP5基因c.3538發生G>A錯義突變,為雜合子;其母FZD4基因c.1482發生G>A無義突變,為雜合子
圖6
基因突變序列圖。6A.FZD4FZD4基因;6B.LRP5基因。患兒c.1482發生G>A無義突變,為雜合子,LRP5基因c.3538發生G>A錯義突變,為雜合子;其父LRP5基因c.3538發生G>A錯義突變,為雜合子;其母FZD4基因c.1482發生G>A無義突變,為雜合子
討論??FEVR是一種以周邊視網膜血管化不完全為臨床特征的遺傳性疾病[1]。其臨床表現、病變過程及遺傳方式呈多樣化,通常同時侵犯雙眼,是導致青少年RD的原因之一[2]。輕度FEVR可無癥狀,然而嚴重的FEVR可致盲[1]。FFA檢查可見患者以及無癥狀的家族成員眼底周邊無血管區,能有效幫助臨床診斷[3]。基因分析可明確診斷[4]。FZD4與LRP5均位于染色體11q,為常染色體顯性遺傳有關基因[5]。FZD4位于11q14.2,在空間結構上可以與Wnt配位結合,從而激活Wnt信號傳導途徑,當FZD4突變時Wnt傳導無法執行其功能,可造成異常血管的發生,或器官組織發生異常[6, 7]。LRP5位于11q13.4,是一個多功能的細胞表面受體,在Wnt信號傳導途徑中,與FZD4結合形成復合體,作為Wnt通路的配體,激活Wnt-beita-catenin信號傳導途徑。LRP5的突變被證實可以導致常染色體隱性遺傳的骨質疏松假神經膠質瘤,以及在人類和鼠模型上的視網膜血管異常[5]。本文報道的2例患兒目前并未出現骨骼等異常,或與年齡較小相關。隨訪中應注意全身疾病的檢查。
本文2例患兒所檢測出的基因突變位點,經與ExAC數據庫中的數據對比,其等位基因頻率均小于1/10 000,我們認為極可能為致病基因突變。例2患兒父親眼底表現正常,而基因檢測中發現LRP5基因發生突變,為雜合子,考慮可能是由于雜合子狀態下的不完全外顯所導致。而每個突變位點是否能單獨致病,需要進一步行突變位點的功能試驗以確認。目前還缺乏FEVR基因型與表現型有明確相關性的報道。FZD4與LRP5突變的FEVR患者臨床表現多樣[5]。但FFA檢查證實所有患者均存在部分視網膜周邊無血管區[1]。然而同時發現兩種基因聯合突變非常少見。Qin等[5]對56個FEVR家系進行了基因測序,結果僅發現一個家系同時存在FZD4和LRP5的突變,且患者癥狀嚴重,提示此兩個基因可能起協同作用。本文報道的2例患兒病變程度較重,均為4期及以上表現,與文獻報道相符。
FEVR是一種少見的遺傳性視網膜血管發育異常,表現多樣,能由多種基因導致。除了眼底照相及FFA檢查,基因檢測具有十分重要的意義,可提示全身疾病以及疾病的嚴重程度。
例1 患兒女,7個月。因發現雙眼斜視3個月就診。出生史G1P1,足月順產,出生體重2900 g;否認吸氧史。骨骼等全身檢查未見異常;新生兒聽力篩查正常。眼科檢查:雙眼眼前節正常。散瞳后應用第三代廣角數碼視網膜成像系統行眼底照相檢查,可見右眼視網膜皺襞連接至晶狀體后伴增生(
例2 患兒女,9歲。因左眼自幼視物不見就診。出生史G1P1,足月順產,出生體重3500 g;否認吸氧史。骨骼等全身檢查未見異常;聽力正常。眼科檢查:右眼視力0.02,左眼視力光感。雙眼眼壓均為13 mmHg(1 mmHg=0.133 kPa)。右眼眼前節正常;左眼晶狀體缺如,其余眼前節正常。眼底檢查,右眼視盤發出的鐮狀皺襞連接至晶狀體后;左眼全RD。眼B型超聲檢查,右眼顳下鐮狀RD伴機化膜;左眼全RD伴玻璃體積血或滲出,眼球壁鈣化(
圖1
雙眼彩色眼底像。1A.右眼,視盤發出的皺襞連至晶狀體后;1B.左眼,視盤血管向顳側牽引伴纖維增生??圖 2?右眼B型超聲像。玻璃體內細帶狀回聲連接視盤至眼前節;玻璃體內機化增生膜與視網膜粘連牽拉,可見部分RD??圖 3?患兒父母FFA像。3A.父親左眼眼底周邊FFA像,血管分支較多,余未見明顯異常;3B.母親左眼周邊FFA像,血管分支較多,走形稍直,其余未見明顯異常??圖 4?右眼B型超聲像。顳下鐮狀RD伴機化膜??圖 5?患兒及父母FFA像。5A, 5B.患兒右眼FFA像,視盤邊界不清,形態異常,向顳側形成一鐮狀RD,其上可見血管;后極部動靜脈充盈尚可,血管僵直,近赤道分支眾多,呈扇形分布,部分血管熒光著染,周邊見近360°無血管區;5C, 5D.母親左右眼周邊FFA像,視網膜血管充盈尚可,血管分支增多,周邊顳側、顳上、顳下均見無血管區;左眼顳下血管末梢吻合,右眼顳下血管少量熒光著染
圖6
基因突變序列圖。6A.FZD4FZD4基因;6B.LRP5基因。患兒c.1482發生G>A無義突變,為雜合子,LRP5基因c.3538發生G>A錯義突變,為雜合子;其父LRP5基因c.3538發生G>A錯義突變,為雜合子;其母FZD4基因c.1482發生G>A無義突變,為雜合子
討論??FEVR是一種以周邊視網膜血管化不完全為臨床特征的遺傳性疾病[1]。其臨床表現、病變過程及遺傳方式呈多樣化,通常同時侵犯雙眼,是導致青少年RD的原因之一[2]。輕度FEVR可無癥狀,然而嚴重的FEVR可致盲[1]。FFA檢查可見患者以及無癥狀的家族成員眼底周邊無血管區,能有效幫助臨床診斷[3]。基因分析可明確診斷[4]。FZD4與LRP5均位于染色體11q,為常染色體顯性遺傳有關基因[5]。FZD4位于11q14.2,在空間結構上可以與Wnt配位結合,從而激活Wnt信號傳導途徑,當FZD4突變時Wnt傳導無法執行其功能,可造成異常血管的發生,或器官組織發生異常[6, 7]。LRP5位于11q13.4,是一個多功能的細胞表面受體,在Wnt信號傳導途徑中,與FZD4結合形成復合體,作為Wnt通路的配體,激活Wnt-beita-catenin信號傳導途徑。LRP5的突變被證實可以導致常染色體隱性遺傳的骨質疏松假神經膠質瘤,以及在人類和鼠模型上的視網膜血管異常[5]。本文報道的2例患兒目前并未出現骨骼等異常,或與年齡較小相關。隨訪中應注意全身疾病的檢查。
本文2例患兒所檢測出的基因突變位點,經與ExAC數據庫中的數據對比,其等位基因頻率均小于1/10 000,我們認為極可能為致病基因突變。例2患兒父親眼底表現正常,而基因檢測中發現LRP5基因發生突變,為雜合子,考慮可能是由于雜合子狀態下的不完全外顯所導致。而每個突變位點是否能單獨致病,需要進一步行突變位點的功能試驗以確認。目前還缺乏FEVR基因型與表現型有明確相關性的報道。FZD4與LRP5突變的FEVR患者臨床表現多樣[5]。但FFA檢查證實所有患者均存在部分視網膜周邊無血管區[1]。然而同時發現兩種基因聯合突變非常少見。Qin等[5]對56個FEVR家系進行了基因測序,結果僅發現一個家系同時存在FZD4和LRP5的突變,且患者癥狀嚴重,提示此兩個基因可能起協同作用。本文報道的2例患兒病變程度較重,均為4期及以上表現,與文獻報道相符。
FEVR是一種少見的遺傳性視網膜血管發育異常,表現多樣,能由多種基因導致。除了眼底照相及FFA檢查,基因檢測具有十分重要的意義,可提示全身疾病以及疾病的嚴重程度。