引用本文: 姚青, 張繼榮, 楊怡, 張茜, 李建寧, 孫玉寧. 沉默信息調節因子1慢病毒表達載體的構建及其在視網膜神經節細胞中的表達. 中華眼底病雜志, 2016, 32(1): 66-69. doi: 10.3760/cma.j.issn.1005-1015.2016.01.016 復制
視網膜神經節細胞(RGC)是投射軸突并形成視神經的神經元,損傷后很難修復或再生[1]。沉默信息調節因子1(sirt1)是依賴于尼克酰胺腺嘌呤二核苷酸(NAD+)的去乙酰化酶,通過去乙酰化作用,使組蛋白和非組蛋白賴氨酸殘基的乙酰基裂解,從而具有調節細胞基因修復、代謝、氧化應激等功能[2, 3]。有研究表明,視神經損傷大鼠的RGC存活數量明顯減少;同時,sirt1 mRNA和蛋白表達水平均下降,并與損傷程度有明顯相關性[4]。但有關sirt1表達降低與RGC損傷的相關機制尚不明確。為此,本研究通過構建sirt1過表達慢病毒感染原代培養的RGC,觀察了感染后RGC中sirt1 mRNA和蛋白表達水平,以期探討sirt1在RGC損傷中的可能作用及機制。現將結果報道如下。
1 材料和方法
慢病毒表達載體pLV5-增強型綠色熒光蛋白(美國SBI公司);限制性內切酶NotⅠ、BamHⅠ(美國NEB公司);脂質體Lipofectamine 2000(LF2000)、Trizol試劑(美國Invitrogen公司);PCR試劑盒(德國Qiagen公司);聚凝胺(美國Sigma公司);大鼠抗sirt1單克隆抗體(美國Abcam公司);蛋白定量試劑盒(美國Pierce公司)。引物合成由北京鼎國昌盛生物有限公司完成。基因測序由上海聯合基因科技有限公司完成。
從GenBank資料庫中獲得大鼠sirt1 mRNA的cDNA序列(序列號XM_006256146.2),根據目的序列和載體多克隆位點,設計并合成引物。sirt1:上游引物5′-CTTAAGCGGCCGCGCCACCATGATTGGCA CCGATCCTC-3′,下游引物5′-GTCGGATCCTCAA TAGTGCTCTGATTTGTCT-3′,擴增片段長度為1680堿基對(bp)。引物中含交換配對堿基、NotⅠ酶切位點、BamHⅠ酶切位點。PCR擴增條件:94℃預變性5 min,94℃變性30 s,54℃退火30 s,72℃延伸1 min 40 s,完成35個循環,4℃保溫。用1%瓊脂糖凝膠電泳分析PCR擴增結果,紫外燈下觀察,切膠,對PCR擴增結果進行純化回收。
取慢病毒表達載體pLV5 5μg及sirt1 PCR擴增產物30μl,用NotⅠ和BamHⅠ雙酶切,總體積40μl,37℃酶切4 h。酶切產物經1%瓊脂糖凝膠電泳后切膠回收。取酶切后的sirt1 PCR擴增片段5μl和pLV5-sirt1載體2μl,用T4連接酶1μl,10倍T4 DNA連接酶緩沖液1μl,室溫放置3 h后,將連接產物轉化到100μl DH5α感受態細胞中,冰上放置30 min,42℃熱激1 min,再放置冰上2 min,加入900μl LB培養液,37℃、100 r/min搖床培養1 h,離心集菌,棄900μl上清液,重懸沉淀,將余下100μl上清液涂于含有氨芐青霉素(100 mmol/L)的LB培養平板上,37℃培養倒置培養12 h。在細菌克隆中挑選合適的菌落進行PCR鑒定,提取陽性克隆的質粒DNA。通過NotⅠ和BamHⅠ雙酶切,驗證構建是否成功。選取7個酶切鑒定正確的克隆進行核酸序列測定,測序結果用DNAstar軟件進行分析。
5%CO2培養箱內以含10%血清的培養液在37℃條件下培養293T細胞24 h,待細胞密度達70%~80%時進行轉染。在無菌的5 ml離心管中加入2.5 ml Opti-MEM培養液,將帶有目的基因的質粒和包裝質粒3:1:1(pLV5-sirt1、pCMV-VSV-G、pCMV-dR8.2)混勻;取另一支無菌的5 ml離心管,加入2.5 ml Opti-MEM培養液,再加入60μl LF2000混勻,混合質粒與LF2000,室溫下溫育5 min,形成質粒與LF2000稀釋液的轉染復合物。將質粒與LF2000混合液轉移至293T細胞的培養液中,混勻,于37℃、5%CO2細胞培養箱中培養。培養8h后倒去含有轉染混和物的培養基,加入含10%血清的細胞培養基,繼續培養48 h。收集293T細胞上清液,濾器過濾后,離心,依據逐孔稀釋滴度法測定慢病毒滴度。
取新生Sprague-Dawley大鼠6只,無菌條件下摘出眼球,鈍性分離視網膜。剪碎視網膜組織,用0.125%的胰蛋白酶消化20 min,細胞沉淀用新鮮培養基培養。將細胞接種至12孔板中的蓋玻片上37℃、5%CO2培養24 h,取出爬片,加入4%多聚甲醛4℃固定10 min;PBS浸洗3次,5 min/次。與異硫氰酸熒光素(FITC)-Thy1.1反應1 h,PBS浸洗3次,5 min/次,將細胞爬片放于載玻片上拍照。以熒光顯微鏡觀察發現的綠色熒光為FITC-Thy1.1陽性,并確認為RGC。接種目的細胞于6 cm培養皿中,分為未感染組、空載體感染組和sirt1過表達慢病毒組。未感染組不作干預;空載體感染組、sirt1過表達慢病毒組以病毒滴度5×108 PFU/ml的空載體慢病毒和sirt1過表達慢病毒分別感染原代培養的大鼠RGC。當細胞融合度達30%~50%時,加入500μl濃縮病毒上清,加入聚凝胺至終濃度8μg/ml,24 h后更換為普通培養液。72 h后,采用熒光顯微鏡觀察發出綠色熒光細胞的平均數與總細胞平均數的比值來確定細胞感染效率。
收集3組的RGC,提取總RNA,反轉錄為cDNA,以磷酸甘油醛脫氫酶(GAPDH)為內參照進行實時PCR反應。引物序列:sirt1:上游引物5′-CCGGACA GCTTCAATAGTG-3′,下游引物5′-CCTGTGGCAG TAACAGTGA C-3′,擴增片段長度249 bp;GAPDH:上游引物5′-ACAGCAACAGGGTGGTGGAC-3′,下游引物5′-TTTGAGGGTGCAGCGAACTT-3′,擴增片段長度252 bp。采用Bio-Rad CF96實時熒光PCR儀進行PCR擴增。
采用蛋白免疫印跡法(Western blot)檢測RGC中sirt1蛋白表達水平。提取未感染組、空載體感染組及sirt1過表達慢病毒組RGC的總蛋白,用二喹啉甲酸蛋白定量試劑盒檢測蛋白濃度,蛋白變性,十二烷基硫酸鈉聚丙烯酰胺凝膠電泳。然后電轉至聚偏氟乙烯膜上,5%脫脂牛奶封閉1.5 h,sirt1一抗(1:1000)4℃孵育過夜,5%脫脂牛奶洗膜,與1:10 000稀釋的羊抗鼠IgG在搖床室溫孵育1 h。增強化學發光法顯色,Image Lab凝膠成像系統成像。以β-肌動蛋白(β-actin)為內參照。對sirt1、β-actin蛋白表達水平進行灰度分析。
各獨立實驗均重復3次。采用SPSS 19.0軟件進行統計分析,計量資料以均數±標準差(
2 結果
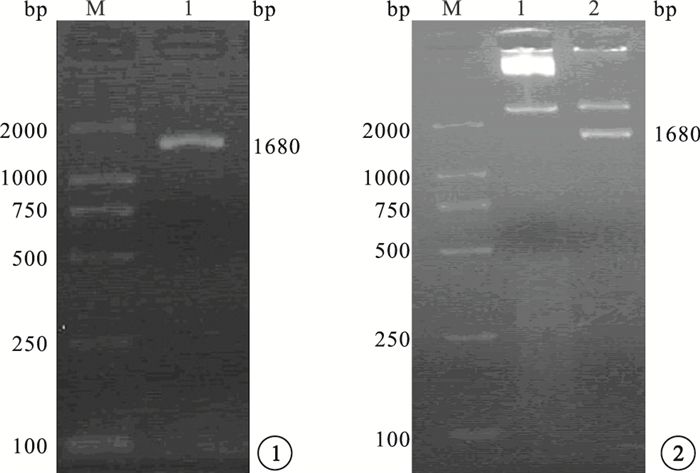
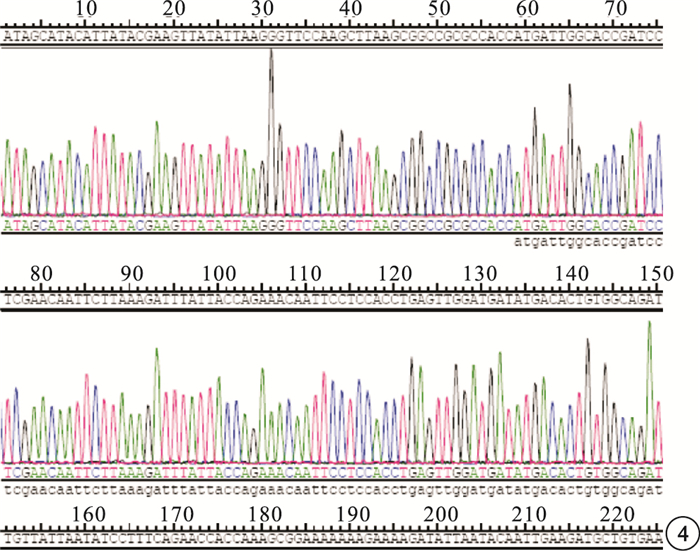
PCR擴增產物凝膠電泳結果顯示,在1680 bp處有擴增條帶,片段大小與預期值一致(圖 1)。使用NotⅠ和BamHⅠ酶切重組質粒pLV5-sirt1,得到約9337 bp的慢病毒載體pLV5片段和1680 bp的sirt1目的基因片段(圖 2)。PCR鑒定結果顯示,表達的DNA條帶與sirt1目的片段大小一致(圖 3)。對重組質粒進行核酸序列測定,與GenBank上sirt1 cDNA標準序列進行對比分析,結果顯示其含有與設計相同的靶序列(圖 4)。
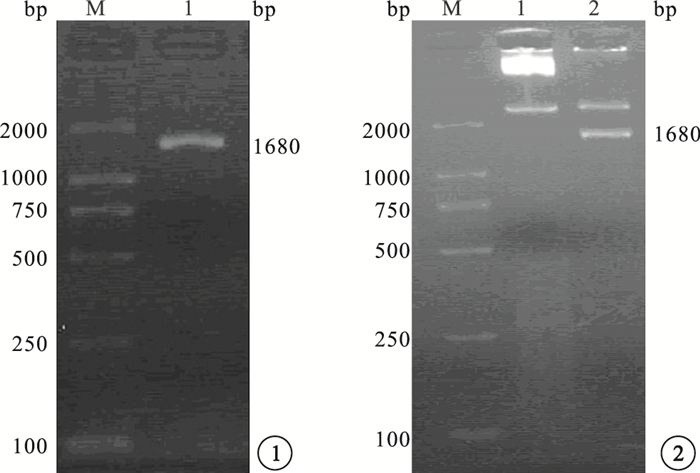 圖1
目的基因sirt1的PCR擴增產物凝膠電泳圖??圖 2 pLV5-sirt1雙酶切鑒定結果
圖1
目的基因sirt1的PCR擴增產物凝膠電泳圖??圖 2 pLV5-sirt1雙酶切鑒定結果
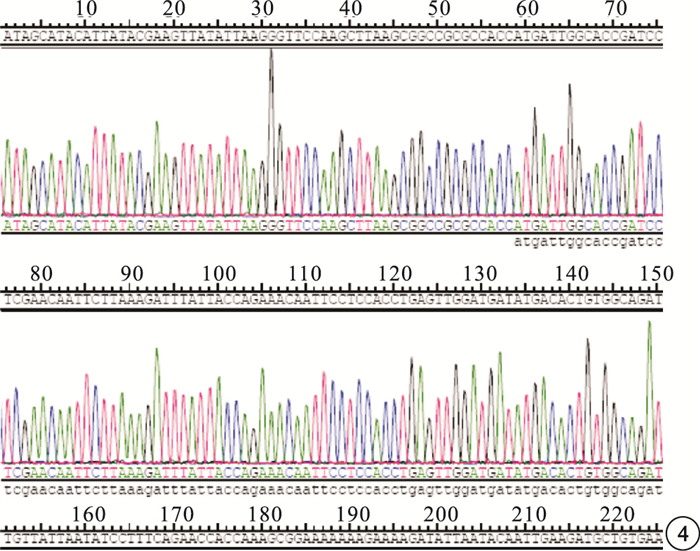 圖3
目的基因sirt1重組克隆PCR鑒定結果
圖3
目的基因sirt1重組克隆PCR鑒定結果
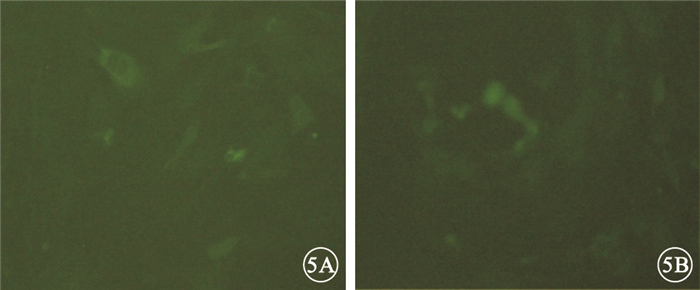 圖4
sirt1慢病毒表達載體pLV5-sirt1的部分測序結果
圖4
sirt1慢病毒表達載體pLV5-sirt1的部分測序結果
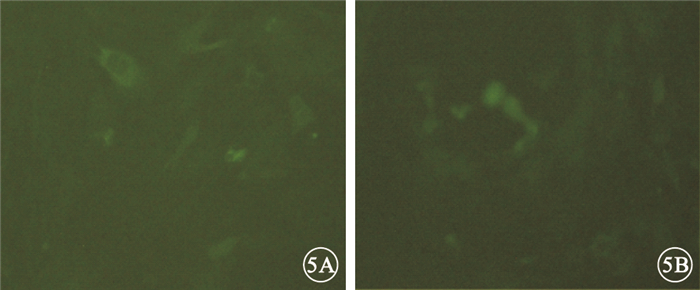
逐孔稀釋滴度法測定結果顯示,病毒經濃縮后的滴度為2×108 PFU/ml。熒光顯微鏡觀察發現,空載體感染組和sirt1過表達慢病毒組均可看到綠色熒光,感染效率在90%以上(圖 5)。
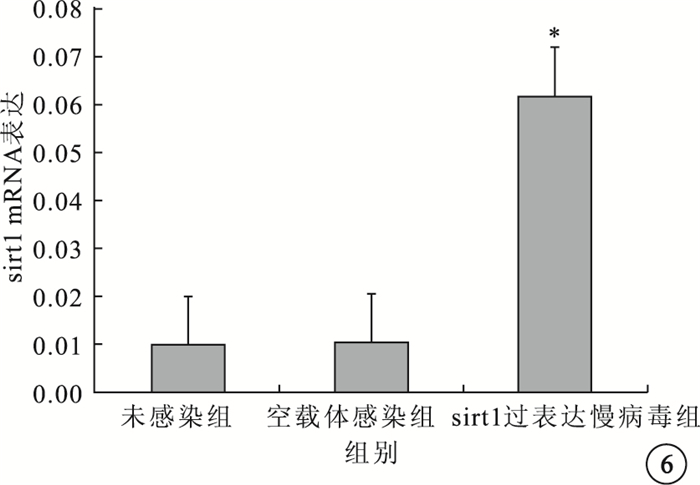 圖5
大鼠RGC熒光顯微鏡像。5A.空載體感染組;5B.sirt1過表達慢病毒組。均可見綠色熒光
圖5
大鼠RGC熒光顯微鏡像。5A.空載體感染組;5B.sirt1過表達慢病毒組。均可見綠色熒光
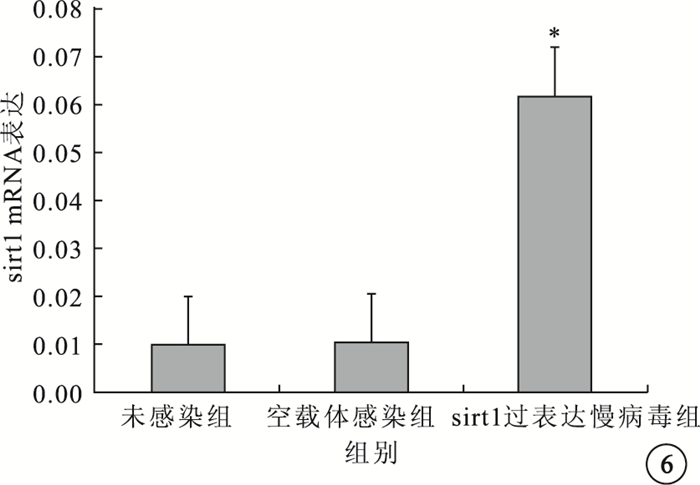
實時PCR檢測結果顯示,sirt1過表達慢病毒組sirt1 mRNA表達水平較未感染組、空載體感染組明顯升高,差異有統計學意義(F=193.5,P<0.05)(圖 6)。
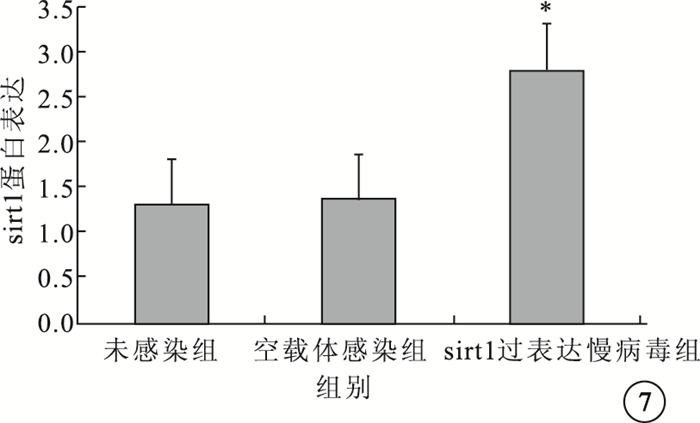 圖6
3組sirt1 mRNA表達水平比較。*與未感染組、空載體感染組比較,P<0.05
圖6
3組sirt1 mRNA表達水平比較。*與未感染組、空載體感染組比較,P<0.05
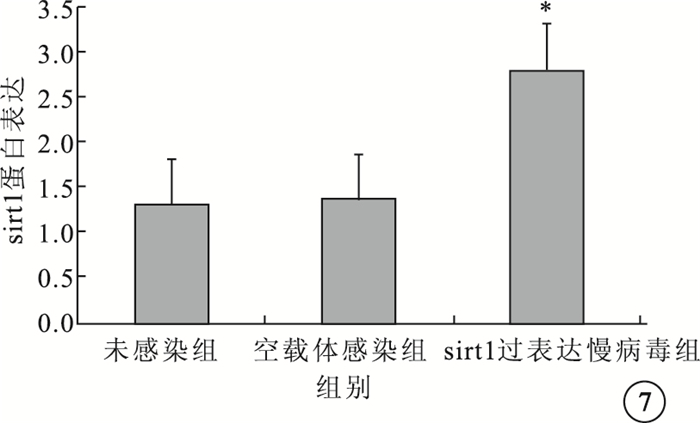
Western blot檢測結果顯示,sirt1過表達慢病毒組sirt1蛋白表達水平較未感染組、空載體感染組明顯升高,差異有統計學意義(F=177.3,P<0.05) (圖 7)。
 圖7
3組sirt1蛋白表達水平比較。*與未感染組、空載體感染組比較,P<0.05
圖7
3組sirt1蛋白表達水平比較。*與未感染組、空載體感染組比較,P<0.05
3 討論
RGC是投射軸突并形成視神經的神經元,能將視覺信息傳遞到視覺中樞。RGC損傷后很難修復或再生[1]。尋找外源性的神經營養物質來預防和治療RGC損傷在視網膜病變的臨床治療方面顯得尤為重要。哺乳動物細胞sirt1是一種依賴于NAD+的去乙酰化酶。在細胞應激狀態下,sirt1可調節某些信號通路以保持細胞生存能力,其中sirt1的去乙酰化酶活性發揮了重要的作用[2, 5, 6]。哺乳動物體內,sirt1定位于細胞核,胚胎早期含量豐富,并在成熟組織中表達廣泛[7]。Sirt1廣泛分布于眼部組織中,在視網膜及視神經退行性變化中起著重要的作用[8]。研究大鼠視神經損傷模型發現,損傷模型大鼠的RGC存活數量明顯減少;同時,sirt1 mRNA和蛋白表達水平均下降,并與損傷的程度有明顯相關性[4]。通過對RGC的研究發現,sirt1的天然激動劑白藜蘆醇可以穩定線粒體的膜電位,下調半胱氨酸蛋白酶,抑制細胞色素C的釋放,從而抑制細胞的凋亡[9, 10]。
慢病毒載體是以人類免疫缺陷Ⅰ型病毒為基礎發展起來的基因治療載體[11]。慢病毒表達載體能有效的將目的基因轉到哺乳動物細胞,而且將慢病毒載體包裝成病毒顆粒,能夠獲得高效轉染的效率[12],轉到靶細胞中的表達載體可以將外源基因有效地整合到宿主染色體上,能夠持久穩定的表達目的基因[13, 14]。
為探討sirt1在RGC中的功能,本研究構建sirt1過表達慢病毒并感染體外原代培養的RGC,觀察sirt1的表達水平。我們首先將sirt1 PCR擴增產物克隆到慢病毒表達載體pLV5,經測序鑒定證明成功構建了pLV5-sirt1過表達慢病毒。在此基礎上,我們利用293T細胞進行了重組慢病毒的包裝,得到了高滴度的sirt1過表達慢病毒。后經pLV5-sirt1過表達慢病毒感染RGC后,利用實時PCR及Western blot對RGC中sirt1的表達水平進行檢測分析。結果證實sirt1過表達慢病毒能明顯提高RGC中sirt1 mRNA及蛋白的表達水平,并且能夠隨著細胞傳代,將sirt1基因穩定地遺傳,為進一步研究sirt1基因的功能及RGC損傷的相互關系奠定了基礎。
視網膜神經節細胞(RGC)是投射軸突并形成視神經的神經元,損傷后很難修復或再生[1]。沉默信息調節因子1(sirt1)是依賴于尼克酰胺腺嘌呤二核苷酸(NAD+)的去乙酰化酶,通過去乙酰化作用,使組蛋白和非組蛋白賴氨酸殘基的乙酰基裂解,從而具有調節細胞基因修復、代謝、氧化應激等功能[2, 3]。有研究表明,視神經損傷大鼠的RGC存活數量明顯減少;同時,sirt1 mRNA和蛋白表達水平均下降,并與損傷程度有明顯相關性[4]。但有關sirt1表達降低與RGC損傷的相關機制尚不明確。為此,本研究通過構建sirt1過表達慢病毒感染原代培養的RGC,觀察了感染后RGC中sirt1 mRNA和蛋白表達水平,以期探討sirt1在RGC損傷中的可能作用及機制。現將結果報道如下。
1 材料和方法
慢病毒表達載體pLV5-增強型綠色熒光蛋白(美國SBI公司);限制性內切酶NotⅠ、BamHⅠ(美國NEB公司);脂質體Lipofectamine 2000(LF2000)、Trizol試劑(美國Invitrogen公司);PCR試劑盒(德國Qiagen公司);聚凝胺(美國Sigma公司);大鼠抗sirt1單克隆抗體(美國Abcam公司);蛋白定量試劑盒(美國Pierce公司)。引物合成由北京鼎國昌盛生物有限公司完成。基因測序由上海聯合基因科技有限公司完成。
從GenBank資料庫中獲得大鼠sirt1 mRNA的cDNA序列(序列號XM_006256146.2),根據目的序列和載體多克隆位點,設計并合成引物。sirt1:上游引物5′-CTTAAGCGGCCGCGCCACCATGATTGGCA CCGATCCTC-3′,下游引物5′-GTCGGATCCTCAA TAGTGCTCTGATTTGTCT-3′,擴增片段長度為1680堿基對(bp)。引物中含交換配對堿基、NotⅠ酶切位點、BamHⅠ酶切位點。PCR擴增條件:94℃預變性5 min,94℃變性30 s,54℃退火30 s,72℃延伸1 min 40 s,完成35個循環,4℃保溫。用1%瓊脂糖凝膠電泳分析PCR擴增結果,紫外燈下觀察,切膠,對PCR擴增結果進行純化回收。
取慢病毒表達載體pLV5 5μg及sirt1 PCR擴增產物30μl,用NotⅠ和BamHⅠ雙酶切,總體積40μl,37℃酶切4 h。酶切產物經1%瓊脂糖凝膠電泳后切膠回收。取酶切后的sirt1 PCR擴增片段5μl和pLV5-sirt1載體2μl,用T4連接酶1μl,10倍T4 DNA連接酶緩沖液1μl,室溫放置3 h后,將連接產物轉化到100μl DH5α感受態細胞中,冰上放置30 min,42℃熱激1 min,再放置冰上2 min,加入900μl LB培養液,37℃、100 r/min搖床培養1 h,離心集菌,棄900μl上清液,重懸沉淀,將余下100μl上清液涂于含有氨芐青霉素(100 mmol/L)的LB培養平板上,37℃培養倒置培養12 h。在細菌克隆中挑選合適的菌落進行PCR鑒定,提取陽性克隆的質粒DNA。通過NotⅠ和BamHⅠ雙酶切,驗證構建是否成功。選取7個酶切鑒定正確的克隆進行核酸序列測定,測序結果用DNAstar軟件進行分析。
5%CO2培養箱內以含10%血清的培養液在37℃條件下培養293T細胞24 h,待細胞密度達70%~80%時進行轉染。在無菌的5 ml離心管中加入2.5 ml Opti-MEM培養液,將帶有目的基因的質粒和包裝質粒3:1:1(pLV5-sirt1、pCMV-VSV-G、pCMV-dR8.2)混勻;取另一支無菌的5 ml離心管,加入2.5 ml Opti-MEM培養液,再加入60μl LF2000混勻,混合質粒與LF2000,室溫下溫育5 min,形成質粒與LF2000稀釋液的轉染復合物。將質粒與LF2000混合液轉移至293T細胞的培養液中,混勻,于37℃、5%CO2細胞培養箱中培養。培養8h后倒去含有轉染混和物的培養基,加入含10%血清的細胞培養基,繼續培養48 h。收集293T細胞上清液,濾器過濾后,離心,依據逐孔稀釋滴度法測定慢病毒滴度。
取新生Sprague-Dawley大鼠6只,無菌條件下摘出眼球,鈍性分離視網膜。剪碎視網膜組織,用0.125%的胰蛋白酶消化20 min,細胞沉淀用新鮮培養基培養。將細胞接種至12孔板中的蓋玻片上37℃、5%CO2培養24 h,取出爬片,加入4%多聚甲醛4℃固定10 min;PBS浸洗3次,5 min/次。與異硫氰酸熒光素(FITC)-Thy1.1反應1 h,PBS浸洗3次,5 min/次,將細胞爬片放于載玻片上拍照。以熒光顯微鏡觀察發現的綠色熒光為FITC-Thy1.1陽性,并確認為RGC。接種目的細胞于6 cm培養皿中,分為未感染組、空載體感染組和sirt1過表達慢病毒組。未感染組不作干預;空載體感染組、sirt1過表達慢病毒組以病毒滴度5×108 PFU/ml的空載體慢病毒和sirt1過表達慢病毒分別感染原代培養的大鼠RGC。當細胞融合度達30%~50%時,加入500μl濃縮病毒上清,加入聚凝胺至終濃度8μg/ml,24 h后更換為普通培養液。72 h后,采用熒光顯微鏡觀察發出綠色熒光細胞的平均數與總細胞平均數的比值來確定細胞感染效率。
收集3組的RGC,提取總RNA,反轉錄為cDNA,以磷酸甘油醛脫氫酶(GAPDH)為內參照進行實時PCR反應。引物序列:sirt1:上游引物5′-CCGGACA GCTTCAATAGTG-3′,下游引物5′-CCTGTGGCAG TAACAGTGA C-3′,擴增片段長度249 bp;GAPDH:上游引物5′-ACAGCAACAGGGTGGTGGAC-3′,下游引物5′-TTTGAGGGTGCAGCGAACTT-3′,擴增片段長度252 bp。采用Bio-Rad CF96實時熒光PCR儀進行PCR擴增。
采用蛋白免疫印跡法(Western blot)檢測RGC中sirt1蛋白表達水平。提取未感染組、空載體感染組及sirt1過表達慢病毒組RGC的總蛋白,用二喹啉甲酸蛋白定量試劑盒檢測蛋白濃度,蛋白變性,十二烷基硫酸鈉聚丙烯酰胺凝膠電泳。然后電轉至聚偏氟乙烯膜上,5%脫脂牛奶封閉1.5 h,sirt1一抗(1:1000)4℃孵育過夜,5%脫脂牛奶洗膜,與1:10 000稀釋的羊抗鼠IgG在搖床室溫孵育1 h。增強化學發光法顯色,Image Lab凝膠成像系統成像。以β-肌動蛋白(β-actin)為內參照。對sirt1、β-actin蛋白表達水平進行灰度分析。
各獨立實驗均重復3次。采用SPSS 19.0軟件進行統計分析,計量資料以均數±標準差(
2 結果
PCR擴增產物凝膠電泳結果顯示,在1680 bp處有擴增條帶,片段大小與預期值一致(圖 1)。使用NotⅠ和BamHⅠ酶切重組質粒pLV5-sirt1,得到約9337 bp的慢病毒載體pLV5片段和1680 bp的sirt1目的基因片段(圖 2)。PCR鑒定結果顯示,表達的DNA條帶與sirt1目的片段大小一致(圖 3)。對重組質粒進行核酸序列測定,與GenBank上sirt1 cDNA標準序列進行對比分析,結果顯示其含有與設計相同的靶序列(圖 4)。
圖1
目的基因sirt1的PCR擴增產物凝膠電泳圖??圖 2 pLV5-sirt1雙酶切鑒定結果
圖3
目的基因sirt1重組克隆PCR鑒定結果
圖4
sirt1慢病毒表達載體pLV5-sirt1的部分測序結果
逐孔稀釋滴度法測定結果顯示,病毒經濃縮后的滴度為2×108 PFU/ml。熒光顯微鏡觀察發現,空載體感染組和sirt1過表達慢病毒組均可看到綠色熒光,感染效率在90%以上(圖 5)。
圖5
大鼠RGC熒光顯微鏡像。5A.空載體感染組;5B.sirt1過表達慢病毒組。均可見綠色熒光
實時PCR檢測結果顯示,sirt1過表達慢病毒組sirt1 mRNA表達水平較未感染組、空載體感染組明顯升高,差異有統計學意義(F=193.5,P<0.05)(圖 6)。
圖6
3組sirt1 mRNA表達水平比較。*與未感染組、空載體感染組比較,P<0.05
Western blot檢測結果顯示,sirt1過表達慢病毒組sirt1蛋白表達水平較未感染組、空載體感染組明顯升高,差異有統計學意義(F=177.3,P<0.05) (圖 7)。
圖7
3組sirt1蛋白表達水平比較。*與未感染組、空載體感染組比較,P<0.05
3 討論
RGC是投射軸突并形成視神經的神經元,能將視覺信息傳遞到視覺中樞。RGC損傷后很難修復或再生[1]。尋找外源性的神經營養物質來預防和治療RGC損傷在視網膜病變的臨床治療方面顯得尤為重要。哺乳動物細胞sirt1是一種依賴于NAD+的去乙酰化酶。在細胞應激狀態下,sirt1可調節某些信號通路以保持細胞生存能力,其中sirt1的去乙酰化酶活性發揮了重要的作用[2, 5, 6]。哺乳動物體內,sirt1定位于細胞核,胚胎早期含量豐富,并在成熟組織中表達廣泛[7]。Sirt1廣泛分布于眼部組織中,在視網膜及視神經退行性變化中起著重要的作用[8]。研究大鼠視神經損傷模型發現,損傷模型大鼠的RGC存活數量明顯減少;同時,sirt1 mRNA和蛋白表達水平均下降,并與損傷的程度有明顯相關性[4]。通過對RGC的研究發現,sirt1的天然激動劑白藜蘆醇可以穩定線粒體的膜電位,下調半胱氨酸蛋白酶,抑制細胞色素C的釋放,從而抑制細胞的凋亡[9, 10]。
慢病毒載體是以人類免疫缺陷Ⅰ型病毒為基礎發展起來的基因治療載體[11]。慢病毒表達載體能有效的將目的基因轉到哺乳動物細胞,而且將慢病毒載體包裝成病毒顆粒,能夠獲得高效轉染的效率[12],轉到靶細胞中的表達載體可以將外源基因有效地整合到宿主染色體上,能夠持久穩定的表達目的基因[13, 14]。
為探討sirt1在RGC中的功能,本研究構建sirt1過表達慢病毒并感染體外原代培養的RGC,觀察sirt1的表達水平。我們首先將sirt1 PCR擴增產物克隆到慢病毒表達載體pLV5,經測序鑒定證明成功構建了pLV5-sirt1過表達慢病毒。在此基礎上,我們利用293T細胞進行了重組慢病毒的包裝,得到了高滴度的sirt1過表達慢病毒。后經pLV5-sirt1過表達慢病毒感染RGC后,利用實時PCR及Western blot對RGC中sirt1的表達水平進行檢測分析。結果證實sirt1過表達慢病毒能明顯提高RGC中sirt1 mRNA及蛋白的表達水平,并且能夠隨著細胞傳代,將sirt1基因穩定地遺傳,為進一步研究sirt1基因的功能及RGC損傷的相互關系奠定了基礎。