引用本文: 薛康, 吳繼紅, 任慧, 張銳, 錢江. 視網膜母細胞瘤低外顯率一家系基因研究. 中華眼底病雜志, 2015, 31(6): 553-555. doi: 10.3760/cma.j.issn.1005-1015.2015.06.010 復制
視網膜母細胞瘤(RB)患者中30%~40%屬于遺傳性RB。根據Knudson[1]兩階段突變假說,RB的形成是由于抑癌基因Rb的雙等位突變或失活。Rb1是首個被克隆的腫瘤抑制基因,定位于13q14,有27個外顯子,DNA全長183 kb,缺乏突變熱點。RB突變數據庫(RBl Gene Mutation Database)目前已收錄超過1200多種突變。RB是單基因遺傳病,多為常染色體顯性遺傳,外顯率高達90%[2];低外顯率RB臨床較為少見,文獻報道尚少。近年出現的外顯子結合目標區域捕獲測序能夠獲得指定區域的遺傳信息[3, 4],極大地提高了Rb基因中外顯子區域的研究效率。我們利用該方法對一個RB低外顯率家系進行了致病基因突變檢測。現將結果報道如下。
1 對象和方法
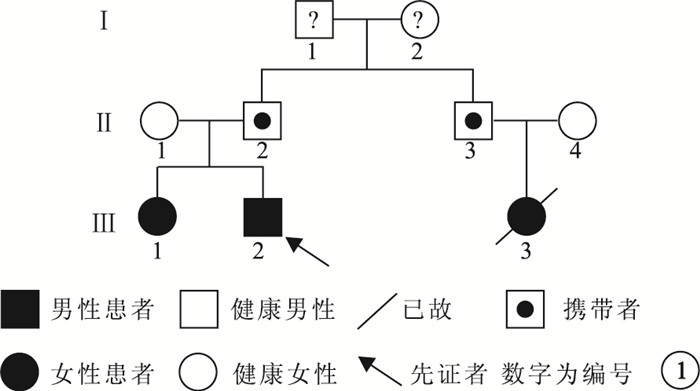
本研究通過復旦大學附屬眼耳鼻喉科醫院倫理審查委員會審查;所有受試者或未成年人監護人均簽署知情同意書。臨床檢查確診的1個RB漢族家系(圖 1)納入研究。先證者(Ⅲ2)為一20個月的男性雙眼RB患兒。詢問家系史時發現患兒家系中尚有另外2例RB患兒。即對該家系進行了臨床及基因檢查。收集患兒既往臨床資料并詳細詢問受試者病史;行雙眼最佳矯正視力、裂隙燈顯微鏡、間接檢眼鏡、眼底彩色照相、眼部B型超聲檢查。RB患兒行第三代廣角數碼視網膜成像系統(RetCamⅢ)眼底照相。家系3代9人中,8人接受眼部臨床檢查;1例RB患兒已死亡。8人中,接受基因檢查6人,其中RB患兒2例。
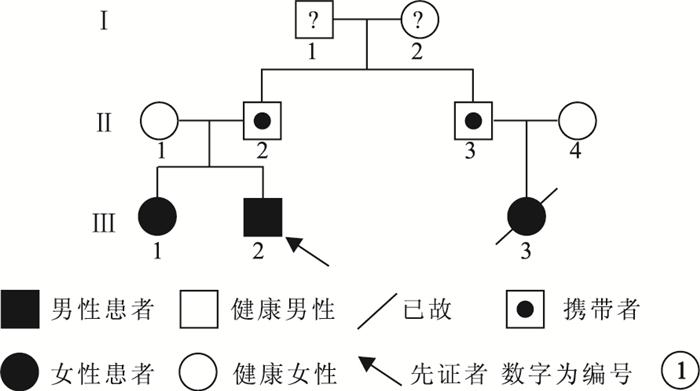 圖1
低外顯率RB家系圖
圖1
低外顯率RB家系圖
采集受試者外周抗凝血5 ml,利用Qiamp Blood試劑盒(德國Qiagen公司)提取外周血全基因組DNA。DNA濃度≥50 mg/L。檢測采用二代高通量測序結合目標區域捕獲技術,對受試者Rb1基因外顯子及其鄰近±10堿基對(bp)內含子區變異進行分析,包括點突變,20 bp以內的缺失插入突變及外顯子水平的拷貝數變異。將基因組DNA隨機打斷為長250~300 bp的片段,在DNA末端標記A,并與Illumina PE接頭一寡核苷酸混合物相連接,連接產物經ligation-mediated聚合酶鏈反應(PCR)擴增、純化,獲得DNA文庫,并對文庫進行質量檢測。將ligation-mediated PCR產物與Rb1靶基因捕獲芯片進行探針雜交、捕獲、富集和洗脫。捕獲的DNA文庫在Illumina HiSeqTM2000平臺(美國Illumina公司)上對目標序列進行高通量測序,得到Illumina原始測序數據。通過IIlumina Basecalling Software 1.7等分析軟件對原始測序數據進行處理得到Raw文件。針對篩選出的突變位點經Sanger驗證排除假陽性,并在所有受試者中驗證是否呈現共分離[5]。
2 結果
3代9人中,RB患者3例。先證者(Ⅲ2)為雙眼RB;Ⅲ1為先證者姐姐,2歲時右眼因RB行眼球摘除。右眼為義眼;左眼眼底、B型超聲檢查,RetCamⅢ眼底照相結果均正常。Ⅲ3為家長自述發現雙眼白瞳,當地醫院確診為雙眼RB,已死亡。其他受試者眼底檢查結果正常。
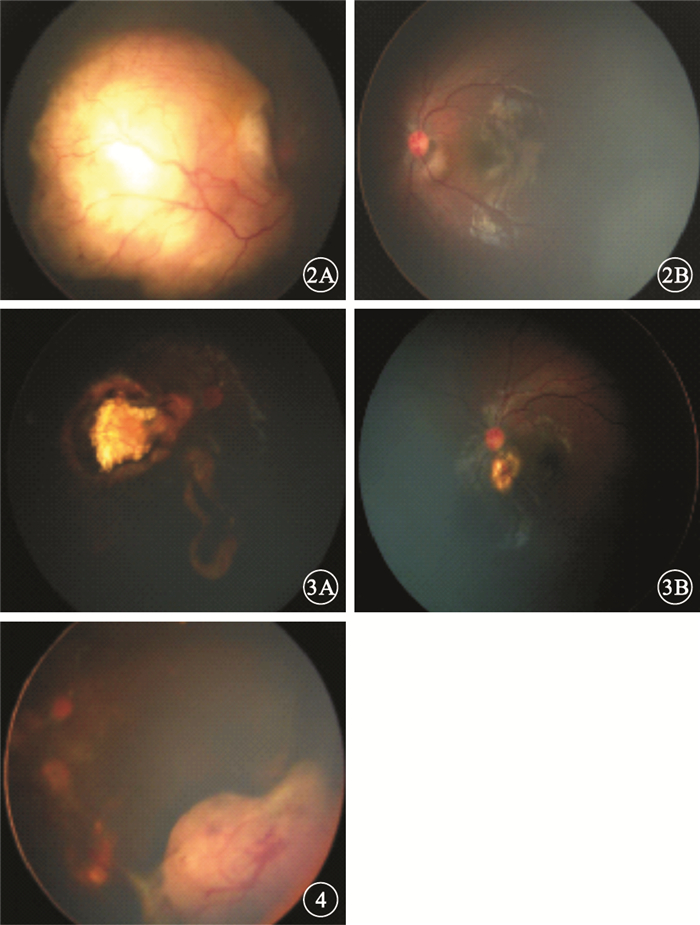
先證者男,20個月。因家長發現患兒右眼發紅、有白色反光就診。RetCamⅢ眼底照相,可見右眼后極部巨大瘤體,伴玻璃體種植、視網膜下積液;左眼視盤旁約1個視盤直徑(DD)大小白色隆起(圖 2)。B型超聲檢查,右眼后極部探及11.5 mm×15.6 mm中高回聲,內有散在鈣化;左眼視盤旁輕度隆起。臨床診斷:雙眼RB;右眼D期,左眼B期。予以8次全身VEC方案化學治療(化療);局部經瞳孔溫熱療法和冷凍治療。治療后右眼瘤體顯著縮小,完全鈣化,呈奶酪-芝士狀消退;左眼瘤體局部萎縮瘢痕化(圖 3)。化療結束后8個月隨訪發現右眼鼻下瘤體增大復發,伴玻璃體種植(圖 4)。予以補充2次化療及全身麻醉下2次冷凍治療。但對再次化療及冷凍治療反應差,遂行右眼眼球摘除手術。病理檢查結果:右眼RB(未分化型)。免疫組織化學檢查結果:波形蛋白(-),低分子量鈣結合蛋白(-),突觸素(+),嗜鉻粒蛋白(-),CD56(+),神經元特異性烯醇化酶(+),結蛋白(+),肌細胞生成素(-),白細胞共同抗原(-),髓過氧化物酶(-),細胞角蛋白(-),Ki-67(50%+)。
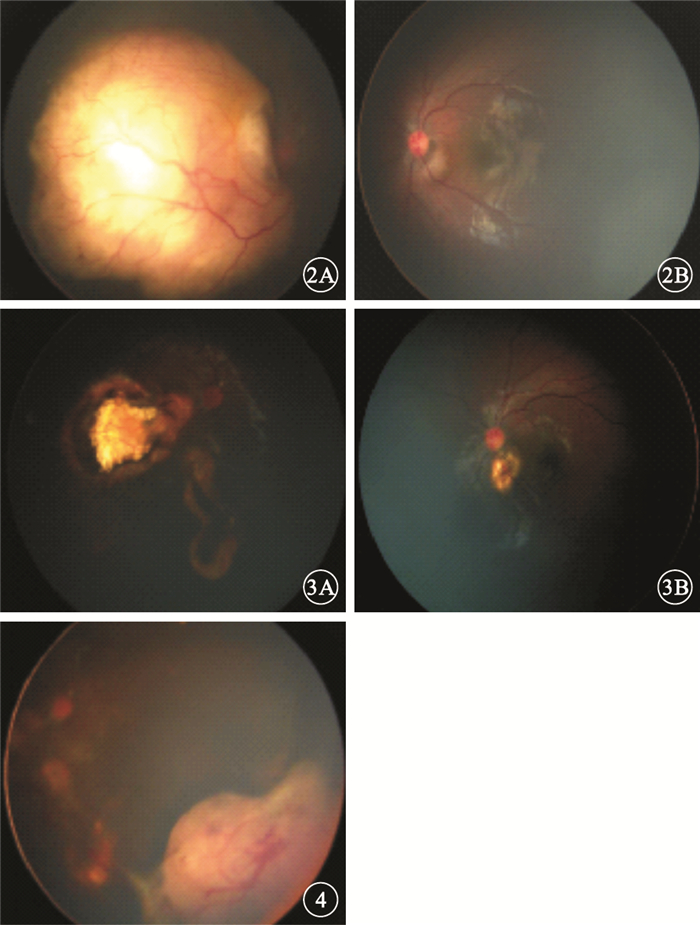 圖2
先證者雙眼彩色眼底像。2A.右眼,后極部巨大瘤體,伴玻璃體種植、視網膜下積液;2B.左眼,視盤旁1 DD大小白色隆起??圖 3先證者雙眼彩色眼底像。3A.右眼,治療后瘤體顯著縮小,完全鈣化;3B.左眼,腫瘤完全消退,表現為脈絡膜瘢痕??圖 4先證者右眼彩色眼底像。鼻下瘤體增大復發
圖2
先證者雙眼彩色眼底像。2A.右眼,后極部巨大瘤體,伴玻璃體種植、視網膜下積液;2B.左眼,視盤旁1 DD大小白色隆起??圖 3先證者雙眼彩色眼底像。3A.右眼,治療后瘤體顯著縮小,完全鈣化;3B.左眼,腫瘤完全消退,表現為脈絡膜瘢痕??圖 4先證者右眼彩色眼底像。鼻下瘤體增大復發
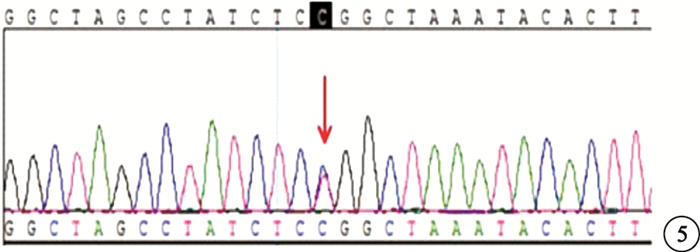
基因檢查結果顯示,先證者(Ⅲ2)的Rb1 NM_000321基因檢出一個錯義突變c.1981C>T (p.Arg661Trp),導致Rb1基因編碼蛋白第661位的精氨酸突變為色氨酸(圖 5)。此突變位點經Sanger驗證排除假陽性。先證者姐姐(Ⅲ1)、先證者父親(Ⅱ2)及其弟弟(Ⅱ3)均存在相同基因位點的突變。Ⅱ2、Ⅱ3配偶(Ⅱ1、Ⅱ4)均未發現該突變。家系中第三代均為RB患兒,第二代均為Rb基因攜帶者,第一代未行基因檢查。推測本家系RB外顯率為50%。
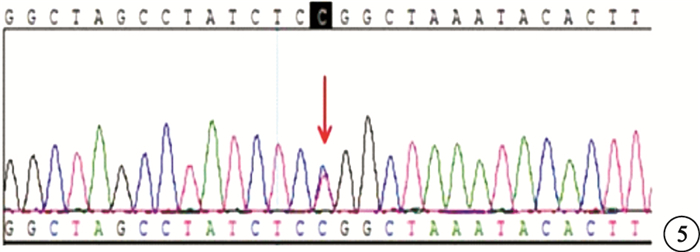 圖5
基因突變序列圖。Rb1 NM_000321基因編碼蛋白第661位的精氨酸突變為色氨酸:c1981 C>T(p.Arg661 Trp)
圖5
基因突變序列圖。Rb1 NM_000321基因編碼蛋白第661位的精氨酸突變為色氨酸:c1981 C>T(p.Arg661 Trp)
3 討論
既往研究結果顯示,遺傳性RB后代RB罹患風險為45%,非遺傳性RB后代RB罹患風險低于1%。遺傳性RB在診斷后40年內二次腫瘤的發生風險為28.00%,接受放療者增加為33.20%,而非遺傳性RB二次腫瘤發生風險僅為1.44%[6]。在化療期間或之后,仍有24%的患者會發生新的RB, 其中絕大部分為早期發病(平均2個月)和遺傳性RB患兒[7]。因此,區別RB患兒是否為遺傳性,有助于估計患兒另一眼發生RB的風險,評估在生命中后期發生第二惡性腫瘤的危險性,預測后代發生RB的風險。在RB患兒及其家庭成員中開展基因診斷和遺傳咨詢具有重要臨床意義。本家系中,先證者(Ⅲ2)在化療結束后產生新的瘤體且治療后仍增大復發,提示遺傳性RB可能需要更為密切和長時間的隨訪。Ⅲ1雖然為單眼RB,但基于基因檢查結果,亦為遺傳性RB,應進行長期密切隨訪。
RB為常染色體顯性遺傳,外顯率高達90%[2]。本家系外顯率為50%。Onadim等[8]對1個低外顯率RB家系的研究結果顯示,該家系所有患者均檢測到錯義突變c.1981C>T,4名無癥狀的家系成員為該突變攜帶者,其余正常成員和38名無血緣關系的健康人中未發現該突變。Abouzeid等[9]在對1個RB家系的研究中發現,家系中有3例雙眼、4例單眼RB患者。對6例在世患者進行基因檢測,發現均攜帶該突變;另外有7名無癥狀家系成員也攜帶該突變,而家系中6名健康人未發現該突變。由此作者認為該突變在該家系中為低外顯突變。Otterson等[10]體外功能試驗結果顯示,p.Arg661Trp突變蛋白仍然具有周期蛋白依賴性激酶介導磷酸化活性,但是該突變蛋白的體外結合功能存在缺陷,這可能是RB低外顯率的原因。提示我們盡管RB的外顯率高達90%, 但仍存在部分低外顯的突變。這種RB中的低外顯現象一方面與錯義突變或框內移碼突變的位置和突變性質有關[11],可表現為pRB蛋白功能的部分缺失;另一方面RB的低外顯現象也與其他遺傳因素的修飾有關[12]。本家系中發現的Rb1 NM_000321基因低外顯的錯義突變c.1981C>T (p.Arg661Trp)為國內首次報道,且RB低外顯率在國內較少報道。本家系中第三代全部外顯發病,而第一代和第二代均未發病,值得臨床工作中注意。而對于家族史陰性的患兒,仍有必要對患兒及其父母進行基因檢測,有助于遺傳咨詢和優生優育。
近年來出現的外顯子結合目標區域測序利用特制的探針針對特定的蛋白編碼區域DNA或某段特定的序列進行捕獲,富集后進行高通量測序,具有效率高、成本低、耗時短的優點,能快速系統地檢測大規模遺傳信息。相比于經典的連鎖分析克隆定位技術,能夠精確篩選出致病基因且不受家系大小的限制,適用于小家系和散發患者。Rushlow等[13]應用定量多重PCR及高靈敏的等位基因特異性PCR技術,對雙眼RB患者的突變檢出率為94.8%。何明燕等[14]采用多個外顯子Sanger法測序,雙眼RB患者突變檢出率為78.57%。本研究存在一定不足,首先為家系報道,病例數有限;其次第一代拒絕行基因檢測,無法明確攜帶者。外顯子結合目標區域測序在RB中的應用報道少見,我們將在今后的工作中在臨床中全面探究外顯子結合目標區域測序在雙眼RB患者中的突變檢出率,并進一步探討,從而比較得出最優RB基因檢測模式,并得以在臨床中推廣應用。
視網膜母細胞瘤(RB)患者中30%~40%屬于遺傳性RB。根據Knudson[1]兩階段突變假說,RB的形成是由于抑癌基因Rb的雙等位突變或失活。Rb1是首個被克隆的腫瘤抑制基因,定位于13q14,有27個外顯子,DNA全長183 kb,缺乏突變熱點。RB突變數據庫(RBl Gene Mutation Database)目前已收錄超過1200多種突變。RB是單基因遺傳病,多為常染色體顯性遺傳,外顯率高達90%[2];低外顯率RB臨床較為少見,文獻報道尚少。近年出現的外顯子結合目標區域捕獲測序能夠獲得指定區域的遺傳信息[3, 4],極大地提高了Rb基因中外顯子區域的研究效率。我們利用該方法對一個RB低外顯率家系進行了致病基因突變檢測。現將結果報道如下。
1 對象和方法
本研究通過復旦大學附屬眼耳鼻喉科醫院倫理審查委員會審查;所有受試者或未成年人監護人均簽署知情同意書。臨床檢查確診的1個RB漢族家系(圖 1)納入研究。先證者(Ⅲ2)為一20個月的男性雙眼RB患兒。詢問家系史時發現患兒家系中尚有另外2例RB患兒。即對該家系進行了臨床及基因檢查。收集患兒既往臨床資料并詳細詢問受試者病史;行雙眼最佳矯正視力、裂隙燈顯微鏡、間接檢眼鏡、眼底彩色照相、眼部B型超聲檢查。RB患兒行第三代廣角數碼視網膜成像系統(RetCamⅢ)眼底照相。家系3代9人中,8人接受眼部臨床檢查;1例RB患兒已死亡。8人中,接受基因檢查6人,其中RB患兒2例。
圖1
低外顯率RB家系圖
采集受試者外周抗凝血5 ml,利用Qiamp Blood試劑盒(德國Qiagen公司)提取外周血全基因組DNA。DNA濃度≥50 mg/L。檢測采用二代高通量測序結合目標區域捕獲技術,對受試者Rb1基因外顯子及其鄰近±10堿基對(bp)內含子區變異進行分析,包括點突變,20 bp以內的缺失插入突變及外顯子水平的拷貝數變異。將基因組DNA隨機打斷為長250~300 bp的片段,在DNA末端標記A,并與Illumina PE接頭一寡核苷酸混合物相連接,連接產物經ligation-mediated聚合酶鏈反應(PCR)擴增、純化,獲得DNA文庫,并對文庫進行質量檢測。將ligation-mediated PCR產物與Rb1靶基因捕獲芯片進行探針雜交、捕獲、富集和洗脫。捕獲的DNA文庫在Illumina HiSeqTM2000平臺(美國Illumina公司)上對目標序列進行高通量測序,得到Illumina原始測序數據。通過IIlumina Basecalling Software 1.7等分析軟件對原始測序數據進行處理得到Raw文件。針對篩選出的突變位點經Sanger驗證排除假陽性,并在所有受試者中驗證是否呈現共分離[5]。
2 結果
3代9人中,RB患者3例。先證者(Ⅲ2)為雙眼RB;Ⅲ1為先證者姐姐,2歲時右眼因RB行眼球摘除。右眼為義眼;左眼眼底、B型超聲檢查,RetCamⅢ眼底照相結果均正常。Ⅲ3為家長自述發現雙眼白瞳,當地醫院確診為雙眼RB,已死亡。其他受試者眼底檢查結果正常。
先證者男,20個月。因家長發現患兒右眼發紅、有白色反光就診。RetCamⅢ眼底照相,可見右眼后極部巨大瘤體,伴玻璃體種植、視網膜下積液;左眼視盤旁約1個視盤直徑(DD)大小白色隆起(圖 2)。B型超聲檢查,右眼后極部探及11.5 mm×15.6 mm中高回聲,內有散在鈣化;左眼視盤旁輕度隆起。臨床診斷:雙眼RB;右眼D期,左眼B期。予以8次全身VEC方案化學治療(化療);局部經瞳孔溫熱療法和冷凍治療。治療后右眼瘤體顯著縮小,完全鈣化,呈奶酪-芝士狀消退;左眼瘤體局部萎縮瘢痕化(圖 3)。化療結束后8個月隨訪發現右眼鼻下瘤體增大復發,伴玻璃體種植(圖 4)。予以補充2次化療及全身麻醉下2次冷凍治療。但對再次化療及冷凍治療反應差,遂行右眼眼球摘除手術。病理檢查結果:右眼RB(未分化型)。免疫組織化學檢查結果:波形蛋白(-),低分子量鈣結合蛋白(-),突觸素(+),嗜鉻粒蛋白(-),CD56(+),神經元特異性烯醇化酶(+),結蛋白(+),肌細胞生成素(-),白細胞共同抗原(-),髓過氧化物酶(-),細胞角蛋白(-),Ki-67(50%+)。
圖2
先證者雙眼彩色眼底像。2A.右眼,后極部巨大瘤體,伴玻璃體種植、視網膜下積液;2B.左眼,視盤旁1 DD大小白色隆起??圖 3先證者雙眼彩色眼底像。3A.右眼,治療后瘤體顯著縮小,完全鈣化;3B.左眼,腫瘤完全消退,表現為脈絡膜瘢痕??圖 4先證者右眼彩色眼底像。鼻下瘤體增大復發
基因檢查結果顯示,先證者(Ⅲ2)的Rb1 NM_000321基因檢出一個錯義突變c.1981C>T (p.Arg661Trp),導致Rb1基因編碼蛋白第661位的精氨酸突變為色氨酸(圖 5)。此突變位點經Sanger驗證排除假陽性。先證者姐姐(Ⅲ1)、先證者父親(Ⅱ2)及其弟弟(Ⅱ3)均存在相同基因位點的突變。Ⅱ2、Ⅱ3配偶(Ⅱ1、Ⅱ4)均未發現該突變。家系中第三代均為RB患兒,第二代均為Rb基因攜帶者,第一代未行基因檢查。推測本家系RB外顯率為50%。
圖5
基因突變序列圖。Rb1 NM_000321基因編碼蛋白第661位的精氨酸突變為色氨酸:c1981 C>T(p.Arg661 Trp)
3 討論
既往研究結果顯示,遺傳性RB后代RB罹患風險為45%,非遺傳性RB后代RB罹患風險低于1%。遺傳性RB在診斷后40年內二次腫瘤的發生風險為28.00%,接受放療者增加為33.20%,而非遺傳性RB二次腫瘤發生風險僅為1.44%[6]。在化療期間或之后,仍有24%的患者會發生新的RB, 其中絕大部分為早期發病(平均2個月)和遺傳性RB患兒[7]。因此,區別RB患兒是否為遺傳性,有助于估計患兒另一眼發生RB的風險,評估在生命中后期發生第二惡性腫瘤的危險性,預測后代發生RB的風險。在RB患兒及其家庭成員中開展基因診斷和遺傳咨詢具有重要臨床意義。本家系中,先證者(Ⅲ2)在化療結束后產生新的瘤體且治療后仍增大復發,提示遺傳性RB可能需要更為密切和長時間的隨訪。Ⅲ1雖然為單眼RB,但基于基因檢查結果,亦為遺傳性RB,應進行長期密切隨訪。
RB為常染色體顯性遺傳,外顯率高達90%[2]。本家系外顯率為50%。Onadim等[8]對1個低外顯率RB家系的研究結果顯示,該家系所有患者均檢測到錯義突變c.1981C>T,4名無癥狀的家系成員為該突變攜帶者,其余正常成員和38名無血緣關系的健康人中未發現該突變。Abouzeid等[9]在對1個RB家系的研究中發現,家系中有3例雙眼、4例單眼RB患者。對6例在世患者進行基因檢測,發現均攜帶該突變;另外有7名無癥狀家系成員也攜帶該突變,而家系中6名健康人未發現該突變。由此作者認為該突變在該家系中為低外顯突變。Otterson等[10]體外功能試驗結果顯示,p.Arg661Trp突變蛋白仍然具有周期蛋白依賴性激酶介導磷酸化活性,但是該突變蛋白的體外結合功能存在缺陷,這可能是RB低外顯率的原因。提示我們盡管RB的外顯率高達90%, 但仍存在部分低外顯的突變。這種RB中的低外顯現象一方面與錯義突變或框內移碼突變的位置和突變性質有關[11],可表現為pRB蛋白功能的部分缺失;另一方面RB的低外顯現象也與其他遺傳因素的修飾有關[12]。本家系中發現的Rb1 NM_000321基因低外顯的錯義突變c.1981C>T (p.Arg661Trp)為國內首次報道,且RB低外顯率在國內較少報道。本家系中第三代全部外顯發病,而第一代和第二代均未發病,值得臨床工作中注意。而對于家族史陰性的患兒,仍有必要對患兒及其父母進行基因檢測,有助于遺傳咨詢和優生優育。
近年來出現的外顯子結合目標區域測序利用特制的探針針對特定的蛋白編碼區域DNA或某段特定的序列進行捕獲,富集后進行高通量測序,具有效率高、成本低、耗時短的優點,能快速系統地檢測大規模遺傳信息。相比于經典的連鎖分析克隆定位技術,能夠精確篩選出致病基因且不受家系大小的限制,適用于小家系和散發患者。Rushlow等[13]應用定量多重PCR及高靈敏的等位基因特異性PCR技術,對雙眼RB患者的突變檢出率為94.8%。何明燕等[14]采用多個外顯子Sanger法測序,雙眼RB患者突變檢出率為78.57%。本研究存在一定不足,首先為家系報道,病例數有限;其次第一代拒絕行基因檢測,無法明確攜帶者。外顯子結合目標區域測序在RB中的應用報道少見,我們將在今后的工作中在臨床中全面探究外顯子結合目標區域測序在雙眼RB患者中的突變檢出率,并進一步探討,從而比較得出最優RB基因檢測模式,并得以在臨床中推廣應用。