引用本文: 張軍燕, 楊秀芬, 顧虹, 徐軍, 劉寧樸, 馬凱. 先天性視網膜劈裂癥一家系基因突變位點分析. 中華眼底病雜志, 2014, 30(1): 62-65. doi: 10.3760/cma.j.issn.1005-1015.2014.01.016 復制
先天性視網膜劈裂癥,又稱X染色體連鎖的青少年型視網膜劈裂癥(XLRS)[1]。Sauer等[2]通過定位克隆的方法將XLRS的致病基因定位于X染色體的短臂遠端Xp22區,稱為RS1基因,包含6個外顯子及5個內含子。該基因編碼一段有224氨基酸的蛋白質稱為視網膜劈裂蛋白。目前已發現的RS1基因突變超過190種,其中多數為突變造成的錯義或無義突變,這部分中又有超過3/4的突變位點位于編碼視網膜劈裂蛋白盤狀結構域的4~6號外顯子;其他突變類型包括剪切位點突變、小的插入或缺失、大片段缺失等。 我們對一個XLRS家系進行了基因突變分析,發現一個新的RS1基因突變位點。現將結果報道如下。
1 對象和方法
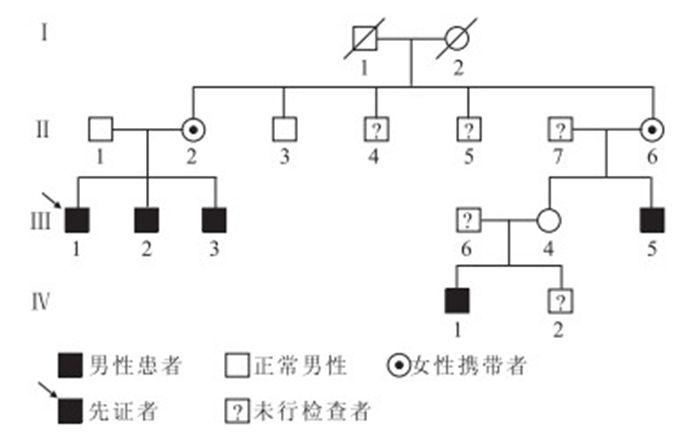
本家系共3代15人(圖 1)。其中,先證者(Ⅲ:1)男,26歲。2009年因左眼自鼻側起視物模糊20 d就診于我院眼科。患者既往有XLRS病史10余年,14年前因右眼孔源性視網膜脫離行右眼玻璃體切割聯合硅油填充手術。家族史中4名男性成員自幼視力差,即對該家系中Ⅱ:2、Ⅱ:3、Ⅱ:6,Ⅲ:2~Ⅲ:5、Ⅳ:1 共計8人16只眼行常規眼部檢查。接受眼部檢查的8人中,男性5人10只眼,女性3人6只眼;年齡9~63歲,平均年齡(37.13±17.81)歲。均行視力、眼壓、裂隙燈顯微鏡、間接檢眼鏡檢查;Ⅲ:2~Ⅲ:5,Ⅳ:1同時行眼底彩色照相和光相干斷層掃描(OCT)檢查。
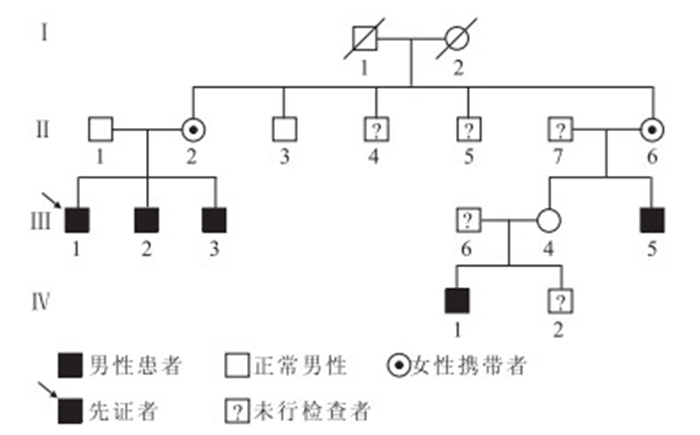 圖1
XLRS家系圖
圖1
XLRS家系圖
本研究經作者所在單位倫理委員會批準并獲得所有受試者知情同意。抽取家系成員中Ⅱ:1~Ⅱ:3、Ⅱ:6、Ⅱ:7,Ⅲ:1~Ⅲ:5,Ⅳ:1、Ⅳ:2 共計12人和社區來源的50名正常對照者外周靜脈血5 ml行RS1基因突變檢測。采用血液基因組DNA提取試劑盒提取DNA(北京天根生物技術公司)。應用聚合酶鏈式反應(PCR)對RS1基因的外顯子1~6進行擴增,引物由上海生工生物工程技術服務有限公司合成(表 1)。使用時濃度稀釋至10 pmol/μl。PCR反應體系總體積為20.0 μl,DNA模板2.0 μl(濃度約為50 ng/μl)、上下游引物各0.5 μl、10倍緩沖液2.0 μl (Mg2+ 濃度為2.0 mmol/L) 、4倍三磷酸脫氧核苷(dNTP) 0.5 μl(dNTP濃度為2.0 mmol/L) 、 DNA聚合酶(Taq)0.5 μl,(Taq濃度為5 U/μl)、去離子水14.0 μl。PCR反應儀(美國Applied Biosystems公司)為96孔熱循環儀,反應程序:預變性94 ℃,5 min;變性94 ℃,30 s,退火58~66 ℃,30 s,延伸72 ℃,45 s,循環35次;最終延伸72 ℃,5 min,4 ℃保存。擴增產物經2%瓊脂糖凝膠電泳,0.5 g/L溴乙錠染色,紫外線光下顯影。所得PCR產物進行雙向直接測序,結果與正常人基因數據庫進行比對定位突變位點。
從Genebank選取6種不同物種,通過CLUSTALOX 1.83軟件對物種間視網膜劈裂蛋白氨基酸序列進行保守性分析。
2 結果
眼前節及眼底檢查正常4人8只眼,其中男性1人(Ⅱ:3)2只眼,女性(Ⅱ:2、Ⅱ:6、Ⅲ:4)3人6只眼。均無視力下降主述。
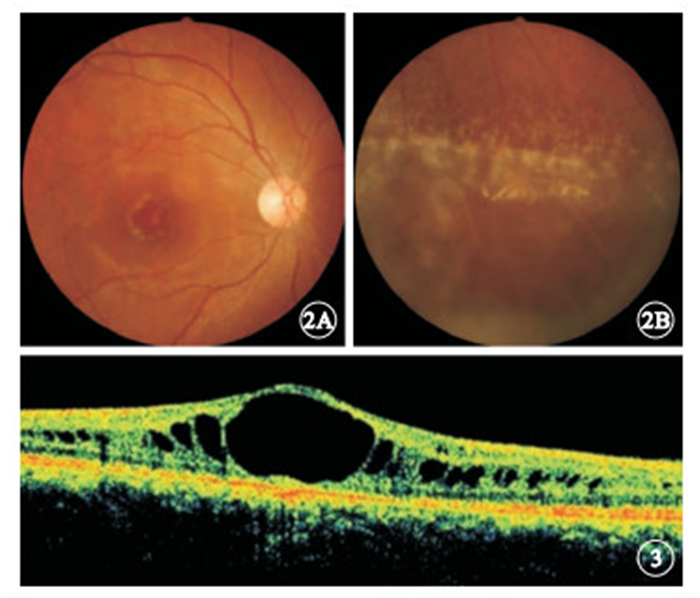
先證者視力右眼眼前手動,左眼0.1,矯正均無提高;眼壓右眼20 mmHg(1 mmHg=0.133 kPa),左眼8 mmHg。右眼晶狀體混濁,眼底不能窺入。左眼眼前節無明顯異常;眼底顳側視網膜脫離,3:00時鐘位方向可見裂孔,周邊視網膜廣泛變性區。后經鞏膜外環扎、放液、外加壓手術,左眼視網膜復位。行眼科常規檢查的家系成員中,Ⅲ:2、Ⅲ:3、Ⅲ:5、Ⅳ:1符合XLRS臨床診斷,均為男性、雙眼受累。患眼視力手動~0.5。出現視力下降時年齡均<10歲。眼底彩色照相可見黃斑區放射狀囊樣劈裂(圖 2A)、周邊視網膜劈裂和白線樣血管、周邊劈裂區囊泡腔消退后白色分界線(圖 2B);OCT檢查可見黃斑區劈裂(圖 3)8只眼,周邊視網膜劈裂6只眼。并發外斜視2只眼、視網膜脫離2只眼、眼球震顫1只眼、玻璃體積血1只眼。
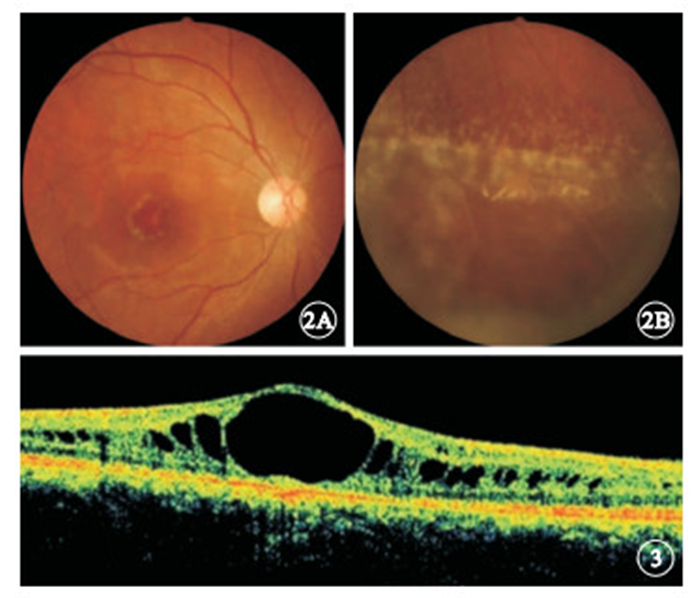 圖2
彩色眼底像。2A.右眼黃斑區劈裂;2B.左眼周邊視網膜劈裂 ? ?圖 3 患眼OCT像。黃斑區劈裂
圖2
彩色眼底像。2A.右眼黃斑區劈裂;2B.左眼周邊視網膜劈裂 ? ?圖 3 患眼OCT像。黃斑區劈裂
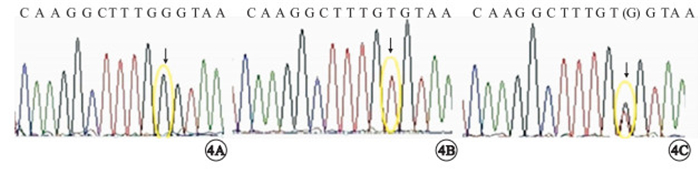
基因突變檢測結果顯示,正常者(4A)3人(Ⅱ:1,Ⅱ:3,Ⅱ:7),均為男性。5例臨床確診者RS1基因在外顯子4的最后一個堿基發生錯義突變(c.326G>T)(圖 4B),導致編碼甘氨酸的密碼子編碼纈氨酸(p.Gly109Val)。Ⅳ:2基因也呈此突變。Ⅳ:2為3歲男性兒童,目前無眼部相關主訴,亦不能配合間接檢眼鏡、OCT等檢查,基因突變檢測結果顯示其具有與5例患者相同的基因突變,遺傳學確定XLRS診斷。眼底檢查正常的3名女性為p.Gly109Val突變的雜合子(圖 4C),為致病基因攜帶者;50名正常對照者未檢出該基因突變。
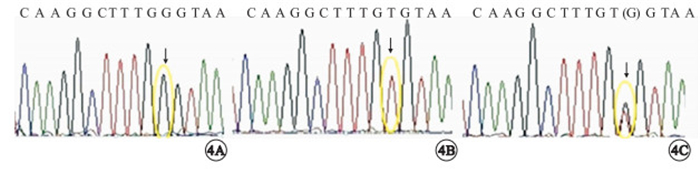 圖4
基因測序圖。4A.家系中眼底檢查正常者RS1基因4號外顯子的末位堿基無突變;4B.患者RS1基因4號外顯子末位堿基發生G>T突變;4C.女性攜帶者該位點為G/T雜合子
圖4
基因測序圖。4A.家系中眼底檢查正常者RS1基因4號外顯子的末位堿基無突變;4B.患者RS1基因4號外顯子末位堿基發生G>T突變;4C.女性攜帶者該位點為G/T雜合子
保守性分析結果顯示,人視網膜劈裂蛋白第109位的甘氨酸在人、家兔、雪貂、鼠中具有相對較高的保守性(圖 5)。
 圖5
視網膜劈裂蛋白77~114位氨基酸序列保守性分析。紅箭處為第109位氨基酸,除牛為谷氨酰胺外,其他物種在該位點均為甘氨酸
圖5
視網膜劈裂蛋白77~114位氨基酸序列保守性分析。紅箭處為第109位氨基酸,除牛為谷氨酰胺外,其他物種在該位點均為甘氨酸
3 討論
本研究為一個三代15人的家系,先證者為青年男性,少年時曾被診斷為XLRS,追溯其家族史,發現家族中多名男性成員自幼視力差,經眼科臨床檢查,確診家系中XLRS患者4例。由于該病為單基因致病,應用PCR聯合直接測序的方法檢測RS1基因,在家系9名成員中檢測出p.Gly109Val突變,確認該突變為致病基因。5例符合臨床診斷的患者均在10歲以前雙眼發病,眼底視網膜黃斑區均有放射樣囊樣劈裂區,但其視力、其他眼科表現均有差異,其中4例分別表現有雙眼視網膜脫離、外斜視、眼球震顫、玻璃體積血等并發癥。表明即使在XLRS的同一家系中,相同基因型患者的臨床表現型可以存在明顯差異。既往的研究結果顯示,XLRS患者的臨床表現具有高度的變異性。一項納入12個中國XLRS家系的研究發現,一部分突變類型導致嚴重型XLRS,而Ser73Pro突變的患者臨床表現相對較輕[3]。與之不同的是,也有研究顯示Ser73Pro突變導致了嚴重型的XLRS表現[4]。一項回顧性研究報道,RS1 的突變類型與患者的視力損害及視網膜電圖(ERG)表現存在一定關聯[5]。XLRS的基因型-表現型關系仍需要更多的研究闡明。
本研究中5例患者RS1基因4號外顯子的末位堿基發生點突變(c.326G>T),導致編碼RS1蛋白的氨基酸序列發生了單個氨基酸的替換p.Gly109Val(GGG→GTG)。家系中3名女性為RS1基因p.Gly109Val突變的雜合子,即為致病基因攜帶者,3人均無視力損害或眼底異常表現。攜帶該病致病基因的女性通常沒有臨床表現,迄今僅見1例病例報道,一位年輕女性具有XLRS的臨床表現和ERG b波改變,該女性從其父親獲得了突變基因,基因型為野生型/突變基因的雜合子;研究者推測其發病原因是由于X染色體失活時,攜帶正常RS1基因的X染色體發生了失活[6]。本研究中除5例患者和3名攜帶者外,還發現家系中1名尚未有臨床表現的3歲男童亦存在p.Gly109Val突變,從遺傳上確立了其XLRS 的診斷,可對其未來病情的發展起到預測作用。
對PubMed、Leiden開放突變數據庫、人類基因突變數據庫(http://www.hgmd.org)中RS1基因的檢 索顯示,p.Gly109Val突變在目前國內外文獻中尚未見報道,為該基因的一個新突變。但已有引起第109位 的甘氨酸替換為谷氨酸、精氨酸、色氨酸(p.Gly109Glu,p.Gly109Arg,p.Gly109Trp)的突變位點的報道[
RS1基因編碼的蛋白質由224個氨基酸組成,稱為視網膜劈裂蛋白,只在視網膜和松果體中表達[2, 11, 12]。在視網膜中,它僅表達于視網膜光感受器細胞和雙極細胞[11]。視網膜劈裂蛋白由4部分組成,N端信號序列,RS1結構域、盤狀結構域以及下游5個氨基酸構成的C端[13]。視網膜劈裂蛋白被認為是一種分泌蛋白,并以八聚體的形式發揮其生理功能,它對維持視網膜正常的結構起著重要作用[14-16]。盤狀結構域是視網膜劈裂蛋白中包含氨基酸最多、最核心的區域,是由8條β鏈構成的盤狀復合體。8條β鏈分為兩組,一組5條,一組3條,分別反向平行地疊連后對接,在此結構基礎上,一端由N端及C端形成的二硫鍵使該復合體結構穩定,另一端則從β鏈形成的復合體結構上伸出3個環,形成溝槽樣腔隙,后者被認為是盤狀結構域與其配體相互識別并結合的位置。另外,在盤狀結構域內部,有兩對半胱氨酸殘端C63-C219和C110-C142,分別構成二硫鍵,這對視網膜劈裂蛋白的正確折疊及穩定性有著至關重要的作用[22]。
有研究指出,位于盤狀結構域的錯義和(或)無義突變主要通過引起蛋白質的錯誤折疊導致視網膜劈裂蛋白功能的喪失。Wang等[17]、Wu和Molday[18]在培養的細胞株上,表達了野生型的視網膜劈裂蛋白以及由盤狀結構域內點突變引起的突變型視網膜劈裂蛋白,比較其表達、生物特性和分泌。結果顯示,野生型的視網膜劈裂蛋白能正常的合成和分泌,而突變型則表現出嚴重的折疊錯誤,并由細胞內控制系統滯留在內質網上,不能被分泌到細胞外發揮其功能。尤其是導致形成二硫鍵的半胱氨酸被替換的突變,產生的蛋白質錯誤折疊并大量凝集。本研究中發現的突變位點p.Gly109Val位于盤狀結構域的第2個環(spike)末端,緊鄰構成分子間二硫鍵的第110位半胱氨酸,因此推測該突變可能是通過引起視網膜劈裂蛋白質折疊錯誤,從而導致視網膜劈裂蛋白功能的喪失。
本研究存在一定的局限性,未能對家系中全部成員進行完整的眼科檢查和ERG等相關輔助檢查,致使臨床資料不夠詳細完整;對RS1基因進行檢測時,正常對照者的樣本量較小,可能導致結果偏倚。但對不同物種間氨基酸保守性的分析仍然支持p.Gly109Val突變為本研究家系的致病位點。作為一個新突變,p.Gly109Val突變對視網膜劈裂蛋白的功能影響機制仍需進一步研究和闡釋。
先天性視網膜劈裂癥,又稱X染色體連鎖的青少年型視網膜劈裂癥(XLRS)[1]。Sauer等[2]通過定位克隆的方法將XLRS的致病基因定位于X染色體的短臂遠端Xp22區,稱為RS1基因,包含6個外顯子及5個內含子。該基因編碼一段有224氨基酸的蛋白質稱為視網膜劈裂蛋白。目前已發現的RS1基因突變超過190種,其中多數為突變造成的錯義或無義突變,這部分中又有超過3/4的突變位點位于編碼視網膜劈裂蛋白盤狀結構域的4~6號外顯子;其他突變類型包括剪切位點突變、小的插入或缺失、大片段缺失等。 我們對一個XLRS家系進行了基因突變分析,發現一個新的RS1基因突變位點。現將結果報道如下。
1 對象和方法
本家系共3代15人(圖 1)。其中,先證者(Ⅲ:1)男,26歲。2009年因左眼自鼻側起視物模糊20 d就診于我院眼科。患者既往有XLRS病史10余年,14年前因右眼孔源性視網膜脫離行右眼玻璃體切割聯合硅油填充手術。家族史中4名男性成員自幼視力差,即對該家系中Ⅱ:2、Ⅱ:3、Ⅱ:6,Ⅲ:2~Ⅲ:5、Ⅳ:1 共計8人16只眼行常規眼部檢查。接受眼部檢查的8人中,男性5人10只眼,女性3人6只眼;年齡9~63歲,平均年齡(37.13±17.81)歲。均行視力、眼壓、裂隙燈顯微鏡、間接檢眼鏡檢查;Ⅲ:2~Ⅲ:5,Ⅳ:1同時行眼底彩色照相和光相干斷層掃描(OCT)檢查。
圖1
XLRS家系圖
本研究經作者所在單位倫理委員會批準并獲得所有受試者知情同意。抽取家系成員中Ⅱ:1~Ⅱ:3、Ⅱ:6、Ⅱ:7,Ⅲ:1~Ⅲ:5,Ⅳ:1、Ⅳ:2 共計12人和社區來源的50名正常對照者外周靜脈血5 ml行RS1基因突變檢測。采用血液基因組DNA提取試劑盒提取DNA(北京天根生物技術公司)。應用聚合酶鏈式反應(PCR)對RS1基因的外顯子1~6進行擴增,引物由上海生工生物工程技術服務有限公司合成(表 1)。使用時濃度稀釋至10 pmol/μl。PCR反應體系總體積為20.0 μl,DNA模板2.0 μl(濃度約為50 ng/μl)、上下游引物各0.5 μl、10倍緩沖液2.0 μl (Mg2+ 濃度為2.0 mmol/L) 、4倍三磷酸脫氧核苷(dNTP) 0.5 μl(dNTP濃度為2.0 mmol/L) 、 DNA聚合酶(Taq)0.5 μl,(Taq濃度為5 U/μl)、去離子水14.0 μl。PCR反應儀(美國Applied Biosystems公司)為96孔熱循環儀,反應程序:預變性94 ℃,5 min;變性94 ℃,30 s,退火58~66 ℃,30 s,延伸72 ℃,45 s,循環35次;最終延伸72 ℃,5 min,4 ℃保存。擴增產物經2%瓊脂糖凝膠電泳,0.5 g/L溴乙錠染色,紫外線光下顯影。所得PCR產物進行雙向直接測序,結果與正常人基因數據庫進行比對定位突變位點。
從Genebank選取6種不同物種,通過CLUSTALOX 1.83軟件對物種間視網膜劈裂蛋白氨基酸序列進行保守性分析。
2 結果
眼前節及眼底檢查正常4人8只眼,其中男性1人(Ⅱ:3)2只眼,女性(Ⅱ:2、Ⅱ:6、Ⅲ:4)3人6只眼。均無視力下降主述。
先證者視力右眼眼前手動,左眼0.1,矯正均無提高;眼壓右眼20 mmHg(1 mmHg=0.133 kPa),左眼8 mmHg。右眼晶狀體混濁,眼底不能窺入。左眼眼前節無明顯異常;眼底顳側視網膜脫離,3:00時鐘位方向可見裂孔,周邊視網膜廣泛變性區。后經鞏膜外環扎、放液、外加壓手術,左眼視網膜復位。行眼科常規檢查的家系成員中,Ⅲ:2、Ⅲ:3、Ⅲ:5、Ⅳ:1符合XLRS臨床診斷,均為男性、雙眼受累。患眼視力手動~0.5。出現視力下降時年齡均<10歲。眼底彩色照相可見黃斑區放射狀囊樣劈裂(圖 2A)、周邊視網膜劈裂和白線樣血管、周邊劈裂區囊泡腔消退后白色分界線(圖 2B);OCT檢查可見黃斑區劈裂(圖 3)8只眼,周邊視網膜劈裂6只眼。并發外斜視2只眼、視網膜脫離2只眼、眼球震顫1只眼、玻璃體積血1只眼。
圖2
彩色眼底像。2A.右眼黃斑區劈裂;2B.左眼周邊視網膜劈裂 ? ?圖 3 患眼OCT像。黃斑區劈裂
基因突變檢測結果顯示,正常者(4A)3人(Ⅱ:1,Ⅱ:3,Ⅱ:7),均為男性。5例臨床確診者RS1基因在外顯子4的最后一個堿基發生錯義突變(c.326G>T)(圖 4B),導致編碼甘氨酸的密碼子編碼纈氨酸(p.Gly109Val)。Ⅳ:2基因也呈此突變。Ⅳ:2為3歲男性兒童,目前無眼部相關主訴,亦不能配合間接檢眼鏡、OCT等檢查,基因突變檢測結果顯示其具有與5例患者相同的基因突變,遺傳學確定XLRS診斷。眼底檢查正常的3名女性為p.Gly109Val突變的雜合子(圖 4C),為致病基因攜帶者;50名正常對照者未檢出該基因突變。
圖4
基因測序圖。4A.家系中眼底檢查正常者RS1基因4號外顯子的末位堿基無突變;4B.患者RS1基因4號外顯子末位堿基發生G>T突變;4C.女性攜帶者該位點為G/T雜合子
保守性分析結果顯示,人視網膜劈裂蛋白第109位的甘氨酸在人、家兔、雪貂、鼠中具有相對較高的保守性(圖 5)。
圖5
視網膜劈裂蛋白77~114位氨基酸序列保守性分析。紅箭處為第109位氨基酸,除牛為谷氨酰胺外,其他物種在該位點均為甘氨酸
3 討論
本研究為一個三代15人的家系,先證者為青年男性,少年時曾被診斷為XLRS,追溯其家族史,發現家族中多名男性成員自幼視力差,經眼科臨床檢查,確診家系中XLRS患者4例。由于該病為單基因致病,應用PCR聯合直接測序的方法檢測RS1基因,在家系9名成員中檢測出p.Gly109Val突變,確認該突變為致病基因。5例符合臨床診斷的患者均在10歲以前雙眼發病,眼底視網膜黃斑區均有放射樣囊樣劈裂區,但其視力、其他眼科表現均有差異,其中4例分別表現有雙眼視網膜脫離、外斜視、眼球震顫、玻璃體積血等并發癥。表明即使在XLRS的同一家系中,相同基因型患者的臨床表現型可以存在明顯差異。既往的研究結果顯示,XLRS患者的臨床表現具有高度的變異性。一項納入12個中國XLRS家系的研究發現,一部分突變類型導致嚴重型XLRS,而Ser73Pro突變的患者臨床表現相對較輕[3]。與之不同的是,也有研究顯示Ser73Pro突變導致了嚴重型的XLRS表現[4]。一項回顧性研究報道,RS1 的突變類型與患者的視力損害及視網膜電圖(ERG)表現存在一定關聯[5]。XLRS的基因型-表現型關系仍需要更多的研究闡明。
本研究中5例患者RS1基因4號外顯子的末位堿基發生點突變(c.326G>T),導致編碼RS1蛋白的氨基酸序列發生了單個氨基酸的替換p.Gly109Val(GGG→GTG)。家系中3名女性為RS1基因p.Gly109Val突變的雜合子,即為致病基因攜帶者,3人均無視力損害或眼底異常表現。攜帶該病致病基因的女性通常沒有臨床表現,迄今僅見1例病例報道,一位年輕女性具有XLRS的臨床表現和ERG b波改變,該女性從其父親獲得了突變基因,基因型為野生型/突變基因的雜合子;研究者推測其發病原因是由于X染色體失活時,攜帶正常RS1基因的X染色體發生了失活[6]。本研究中除5例患者和3名攜帶者外,還發現家系中1名尚未有臨床表現的3歲男童亦存在p.Gly109Val突變,從遺傳上確立了其XLRS 的診斷,可對其未來病情的發展起到預測作用。
對PubMed、Leiden開放突變數據庫、人類基因突變數據庫(http://www.hgmd.org)中RS1基因的檢 索顯示,p.Gly109Val突變在目前國內外文獻中尚未見報道,為該基因的一個新突變。但已有引起第109位 的甘氨酸替換為谷氨酸、精氨酸、色氨酸(p.Gly109Glu,p.Gly109Arg,p.Gly109Trp)的突變位點的報道[
RS1基因編碼的蛋白質由224個氨基酸組成,稱為視網膜劈裂蛋白,只在視網膜和松果體中表達[2, 11, 12]。在視網膜中,它僅表達于視網膜光感受器細胞和雙極細胞[11]。視網膜劈裂蛋白由4部分組成,N端信號序列,RS1結構域、盤狀結構域以及下游5個氨基酸構成的C端[13]。視網膜劈裂蛋白被認為是一種分泌蛋白,并以八聚體的形式發揮其生理功能,它對維持視網膜正常的結構起著重要作用[14-16]。盤狀結構域是視網膜劈裂蛋白中包含氨基酸最多、最核心的區域,是由8條β鏈構成的盤狀復合體。8條β鏈分為兩組,一組5條,一組3條,分別反向平行地疊連后對接,在此結構基礎上,一端由N端及C端形成的二硫鍵使該復合體結構穩定,另一端則從β鏈形成的復合體結構上伸出3個環,形成溝槽樣腔隙,后者被認為是盤狀結構域與其配體相互識別并結合的位置。另外,在盤狀結構域內部,有兩對半胱氨酸殘端C63-C219和C110-C142,分別構成二硫鍵,這對視網膜劈裂蛋白的正確折疊及穩定性有著至關重要的作用[22]。
有研究指出,位于盤狀結構域的錯義和(或)無義突變主要通過引起蛋白質的錯誤折疊導致視網膜劈裂蛋白功能的喪失。Wang等[17]、Wu和Molday[18]在培養的細胞株上,表達了野生型的視網膜劈裂蛋白以及由盤狀結構域內點突變引起的突變型視網膜劈裂蛋白,比較其表達、生物特性和分泌。結果顯示,野生型的視網膜劈裂蛋白能正常的合成和分泌,而突變型則表現出嚴重的折疊錯誤,并由細胞內控制系統滯留在內質網上,不能被分泌到細胞外發揮其功能。尤其是導致形成二硫鍵的半胱氨酸被替換的突變,產生的蛋白質錯誤折疊并大量凝集。本研究中發現的突變位點p.Gly109Val位于盤狀結構域的第2個環(spike)末端,緊鄰構成分子間二硫鍵的第110位半胱氨酸,因此推測該突變可能是通過引起視網膜劈裂蛋白質折疊錯誤,從而導致視網膜劈裂蛋白功能的喪失。
本研究存在一定的局限性,未能對家系中全部成員進行完整的眼科檢查和ERG等相關輔助檢查,致使臨床資料不夠詳細完整;對RS1基因進行檢測時,正常對照者的樣本量較小,可能導致結果偏倚。但對不同物種間氨基酸保守性的分析仍然支持p.Gly109Val突變為本研究家系的致病位點。作為一個新突變,p.Gly109Val突變對視網膜劈裂蛋白的功能影響機制仍需進一步研究和闡釋。