引用本文: 程萌, 楊鴿, 雷博, 劉宇瑩, 金學民. Usher綜合征2型一家系USH2A基因新突變. 中華眼底病雜志, 2018, 34(3): 268-271. doi: 10.3760/cma.j.issn.1005-1015.2018.03.014 復制
Usher綜合征(USH)是一種以耳聾和視網膜色素變性(RP)為特征的常染色體隱性遺傳疾病[1]。臨床上根據聽力和前庭功能受累情況將其分為1(USH1)、2(USH2)、3(USH3)等3種臨床亞型[2]。其中,USH2最為常見[3],表現為先天性中度-重度非漸進性耳聾,前庭功能正常,20歲左右出現RP癥狀[4, 5]。USH具有高度遺傳異質性,研究發現,超過70%的USH2是USH2A基因突變所致[6]。我們對一個USH2家系進行了相關致病基因檢測,旨在明確該家系患者的致病基因。現將結果報道如下。
1 對象和方法
本研究獲鄭州大學第一附屬醫院倫理委員會批準;嚴格遵守赫爾辛基宣言,所有受試者及未成年受試者監護人均簽署知情同意書。一個三代USH2家系(圖1)中2例患者及5名正常家系成員納入研究。所有受試者均行最佳矯正視力(BCVA)、裂隙燈顯微鏡、間接檢眼鏡、全視野視網膜電圖(ERG)、光相干斷層掃描(OCT)、視野檢查,以及前庭功能和純音測聽檢查。同時招募100名無血緣關系的健康志愿者作為正常對照者進行USH基因測序比對。
先證者(Ⅱ2),男,32歲。10歲時雙耳出現聽力下降;目前雙耳重度感音神經性耳聾,前庭功能未見異常。12歲時雙眼出現夜盲。雙眼BCVA分別為0.4、0.7。視盤顏色淡,視網膜血管變細,周邊彌漫性骨細胞樣色素顆粒沉著(圖2)。全視野ERG明適應、暗適應a、b波重度下降。OCT檢查可見黃斑中心凹以外區域視網膜明顯變薄,尤以視細胞層明顯(圖3)。視野向心性縮小(圖4)。先證者弟弟(Ⅱ3),30歲。雙耳重度感音神經性耳聾,前庭功能正常。雙眼BCVA分別為0.9、0.8。眼底表現及全視野ERG、OCT、視野檢查結果同先證者(Ⅱ2)。家系成員BCVA、眼底、視野、OCT、全視野ERG檢查均未見異常。依據臨床檢查結果并結合家系分析,患者符合常染色體隱性遺傳USH2診斷標準[7]。
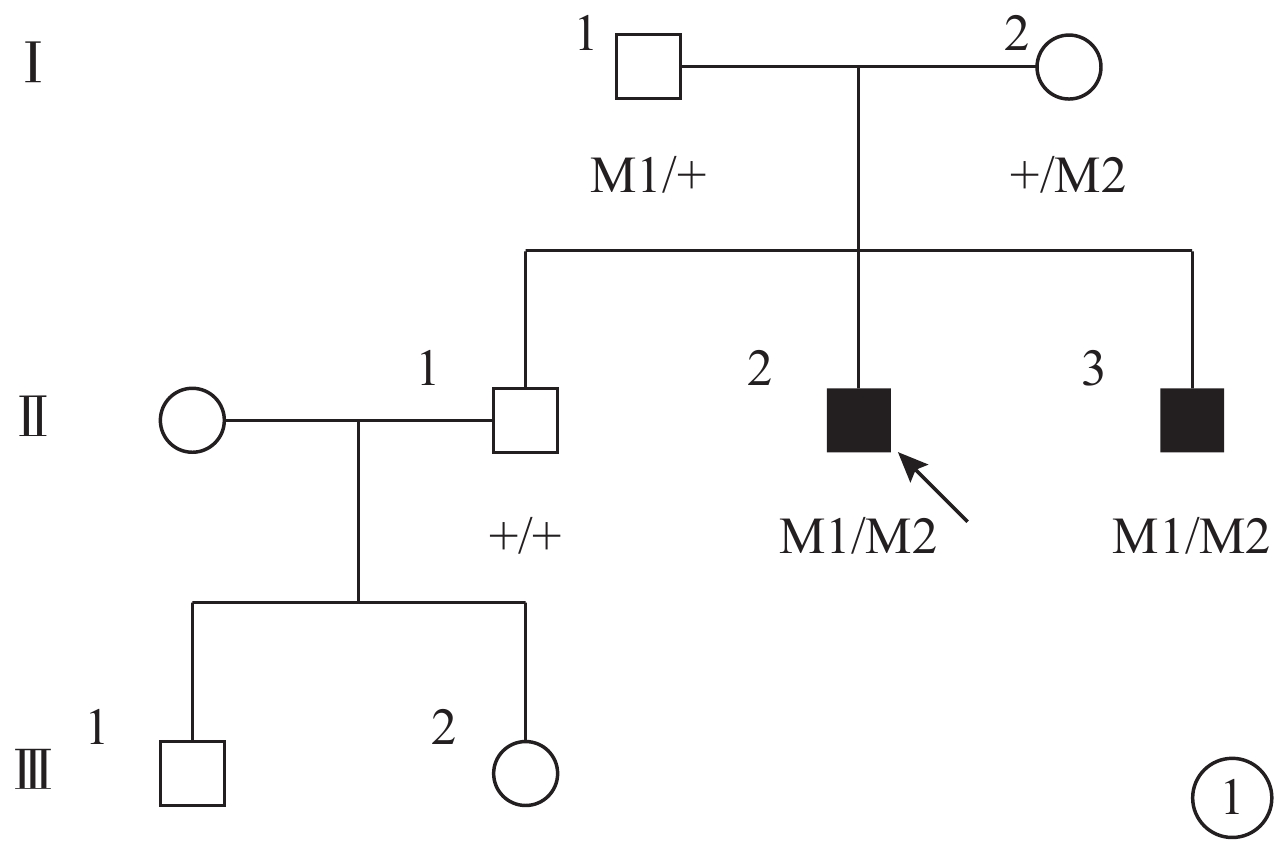
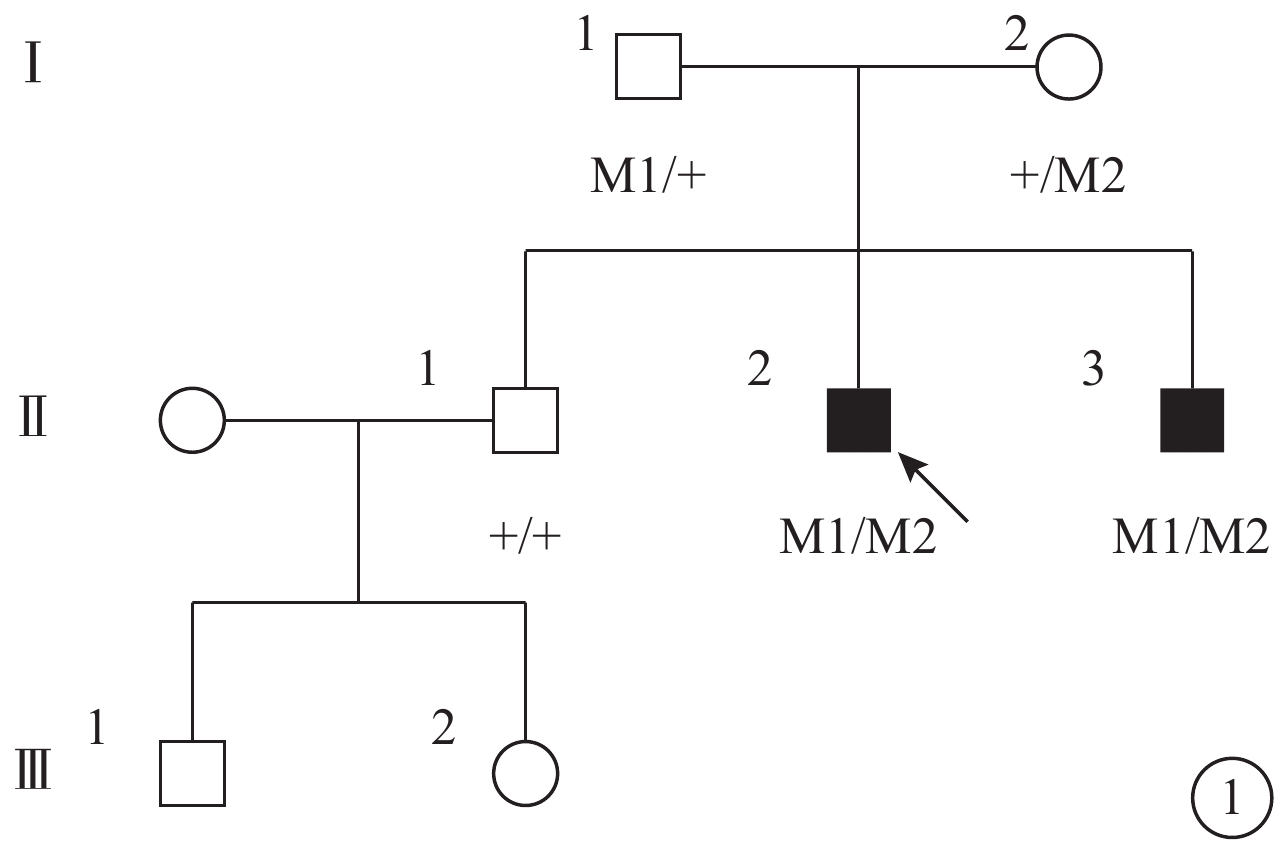 圖1
患者家系圖。■:男性患者;□:正常男性;○:正常女性;↑:先證者;M1:p.I397K/c.1190T>A;M2:p.M182T/c.5459T>C;M3:G268R/c.802G>A
圖1
患者家系圖。■:男性患者;□:正常男性;○:正常女性;↑:先證者;M1:p.I397K/c.1190T>A;M2:p.M182T/c.5459T>C;M3:G268R/c.802G>A
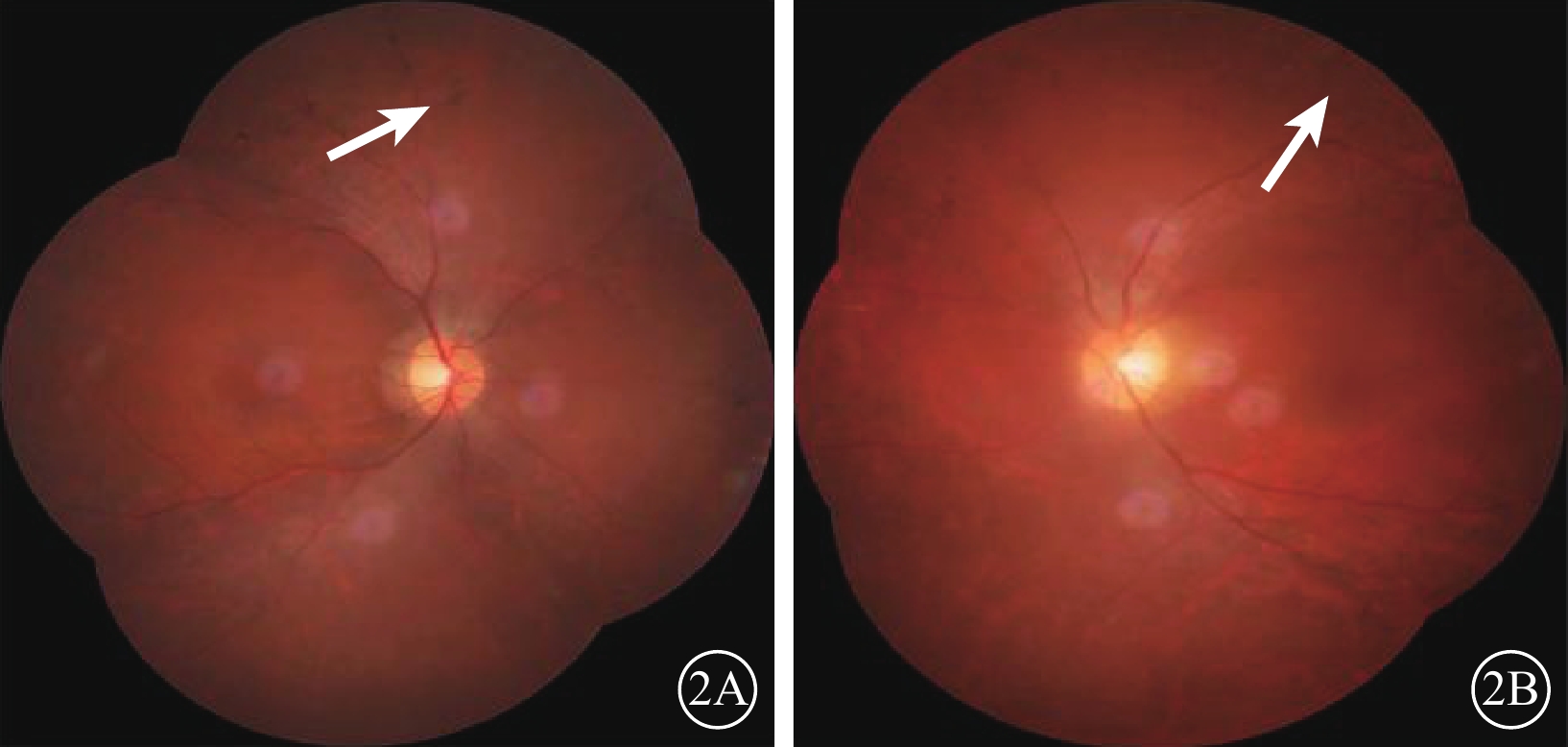 圖2
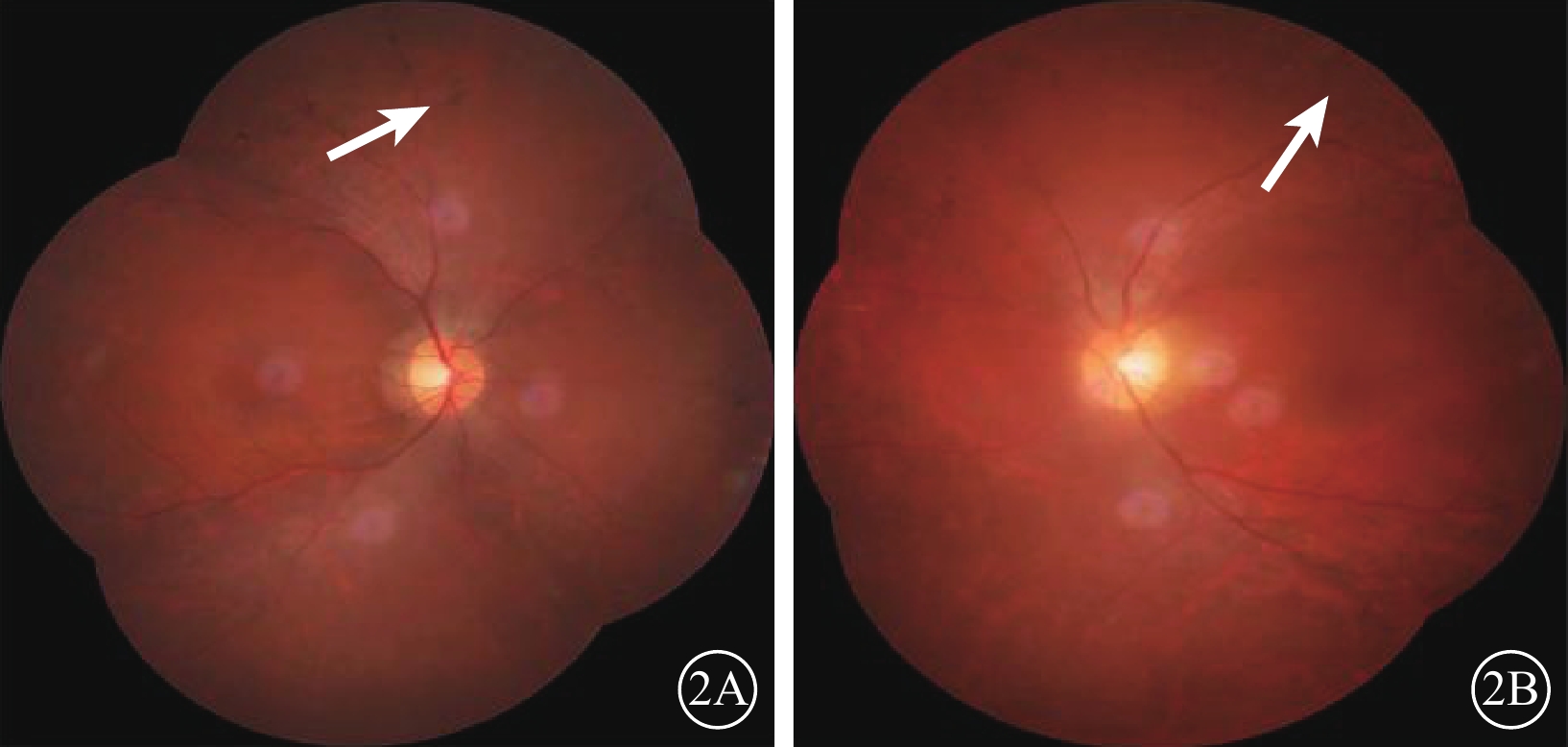
先證者(Ⅱ2)雙眼彩色眼底像。2A. 右眼;2B. 左眼。視盤顏色淡,視網膜血管變細;周邊部視網膜骨細胞樣色素沉著(白箭)
圖2
先證者(Ⅱ2)雙眼彩色眼底像。2A. 右眼;2B. 左眼。視盤顏色淡,視網膜血管變細;周邊部視網膜骨細胞樣色素沉著(白箭)
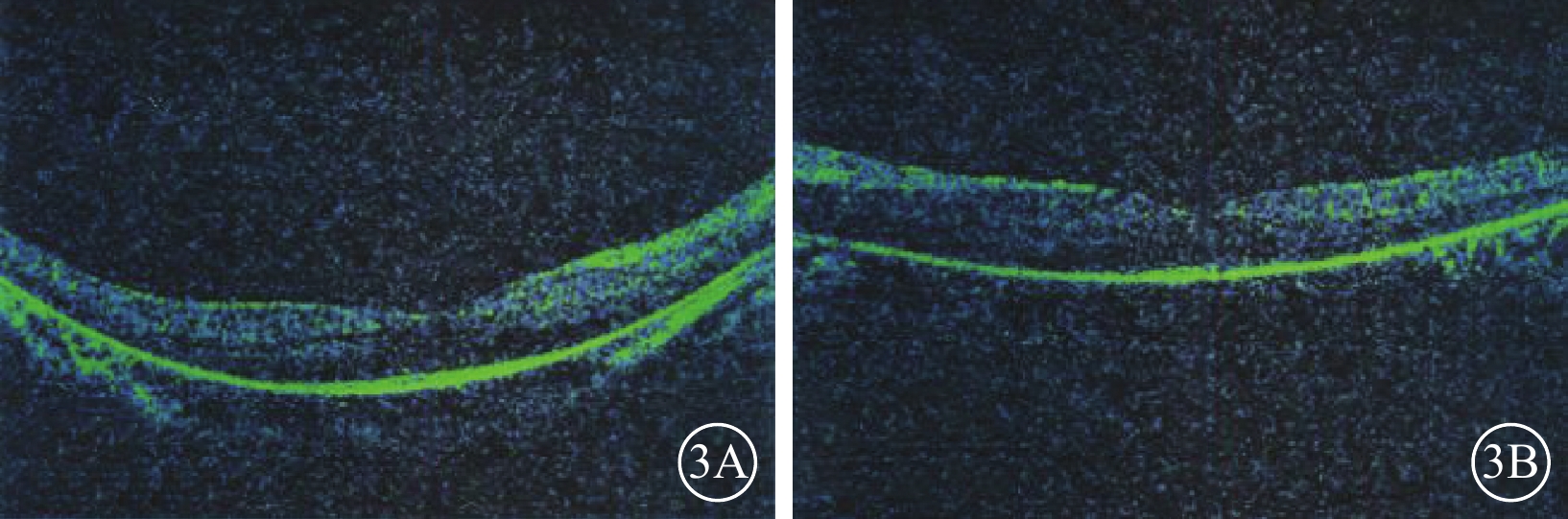 圖3
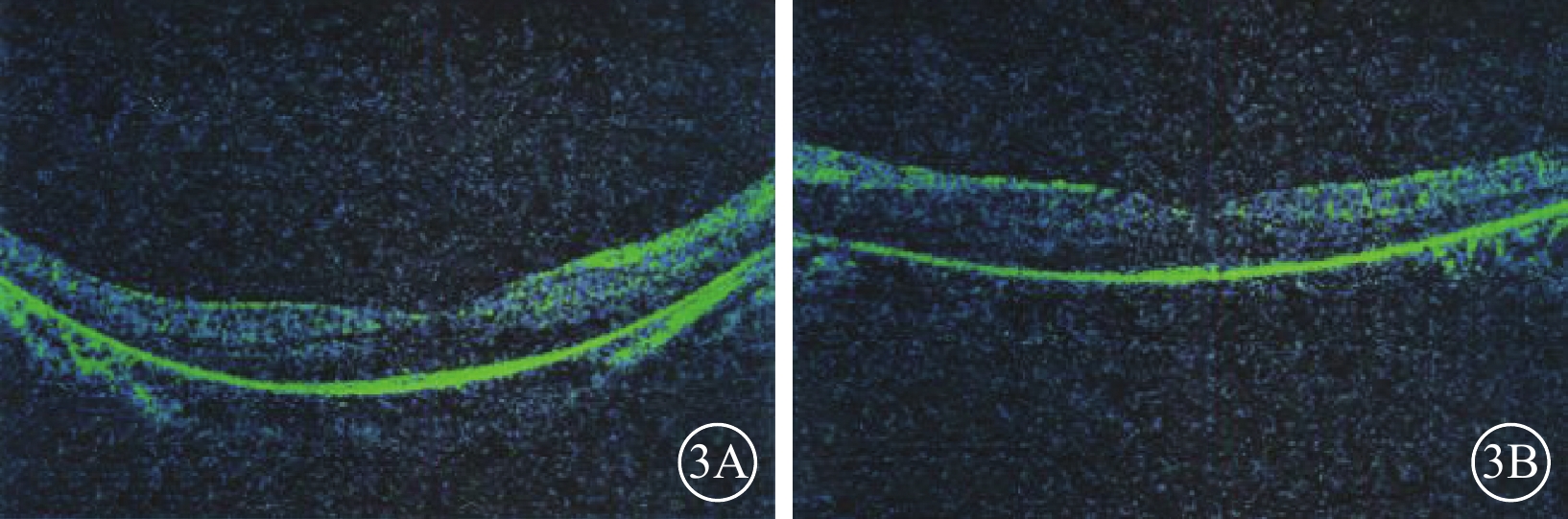
先證者(Ⅱ2)雙眼OCT像。3A. 右眼;3B. 左眼。黃斑中心凹以外區域視網膜明顯變薄,尤其以視細胞層更為明顯
圖3
先證者(Ⅱ2)雙眼OCT像。3A. 右眼;3B. 左眼。黃斑中心凹以外區域視網膜明顯變薄,尤其以視細胞層更為明顯
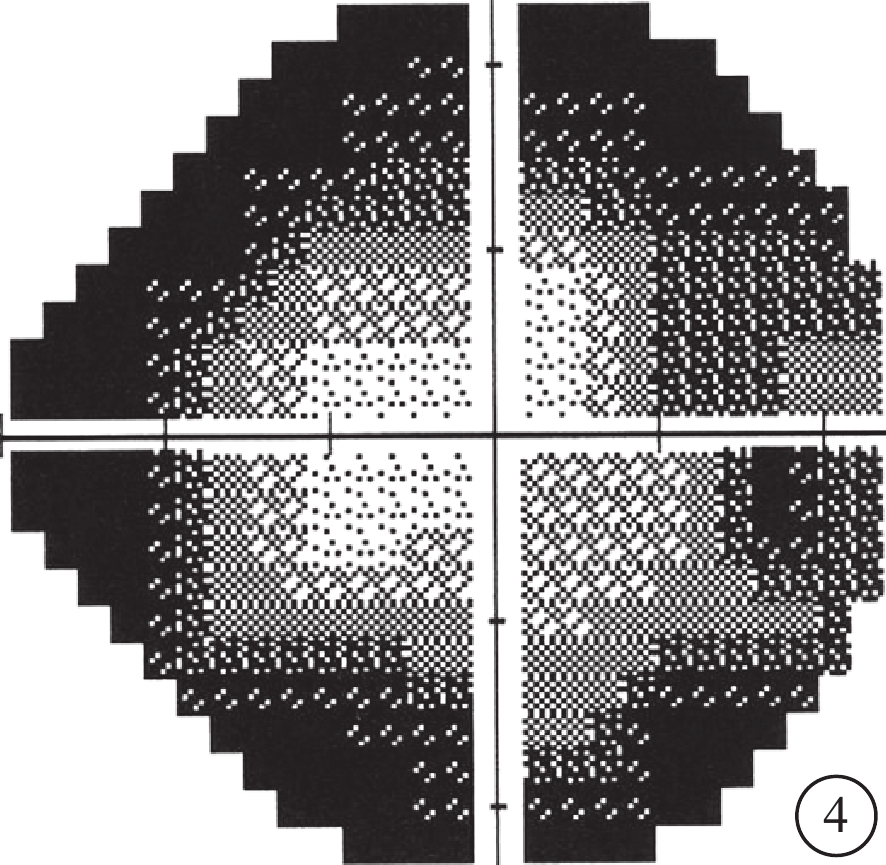 圖4
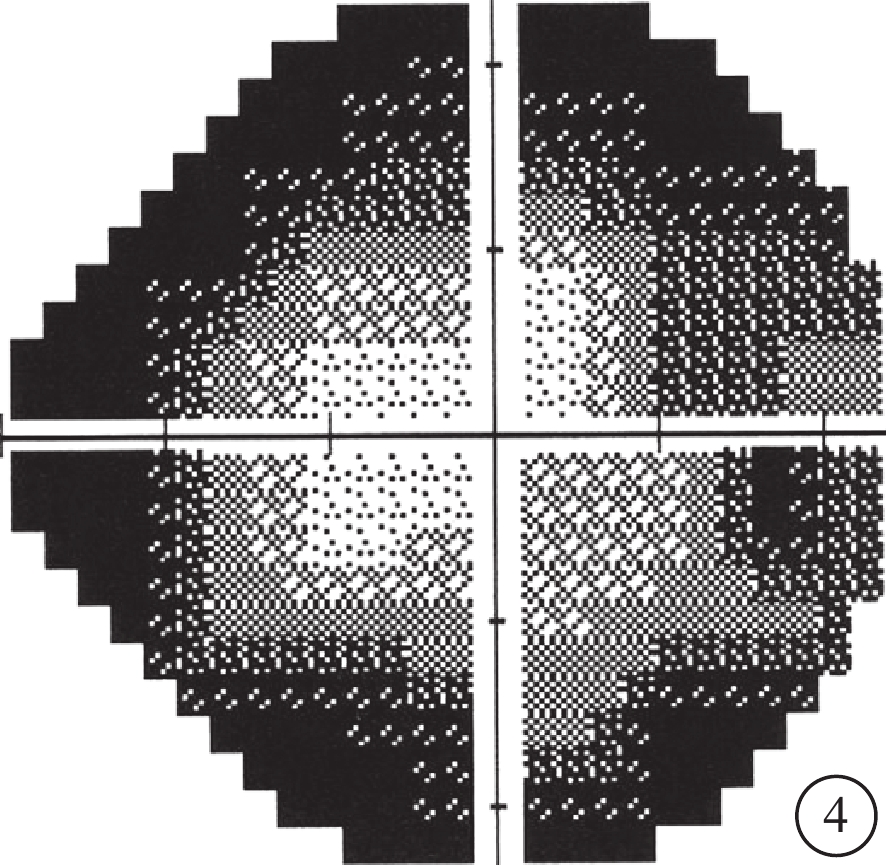
先證者(Ⅱ2)視野檢查像。視野向心性縮小
圖4
先證者(Ⅱ2)視野檢查像。視野向心性縮小
采集家系中7名受試者的外周靜脈血3 ml,乙二胺四乙酸抗凝。采用天根生化科技(北京)有限公血液基因組DNA提取試劑盒按照標準操作流程提取全基因組DNA,紫外分光光度計定量。應用北京邁基諾公司提供的遺傳眼病目標基因試劑盒對目前已知的136個遺傳性視網膜疾病致病基因的外顯子區域進行捕獲。每例患者提取3 μg DNA作為起始量,采用超聲儀進行超聲片段化,打斷樣本片段為150堿基對,對其進行末端補平、兩端加A、連接adoptor、純化,文庫質量檢測后進行雜交、洗脫、連接介導聚合酶鏈反應(PCR)擴增對捕獲文庫富集。
PCR產物采用Beckman Ampure beads按1.8:1體積進行純化,應用II-lumina HiSeq 2500進行高通量測序。測序結果通過基因組數據庫、共識編碼序列計劃、NCBI數據庫單核苷酸多態性(SNP)子庫、單核苷酸多態性數據庫、千人基因組計劃、人基因組數據庫對SNP進行注釋,確定候選致病基因突變位點。
所有檢測出的可能致病突變均通過Sanger測序進行驗證并進行家系共分離驗證。Sanger測序結果再通過SIFT預測去掉不影響蛋白功能的SNPs,最終確定致病性基因突變位點。
2 結果
USH基因測序結果顯示,2例患者(Ⅱ2、Ⅱ3)的USH2A基因第27、7、5號外顯子分別存在c.5459T>C(p.M1820T)、c.1190T>A(p.I397K)、c.802G>A(p.G268R)3個雜合性錯義突變(圖5)。其中,27號外顯子第5 459位核糖核苷酸T變C,導致其編碼的第1820位氨基酸由甲硫氨酸變為蘇氨酸;7號外顯子第1190位核糖核苷酸T變A,導致其編碼的第397位氨基酸由異亮氨酸變為賴氨酸;5號外顯子第802位核糖核苷酸G變A,導致其編碼的第268位氨基酸由甘氨酸變為精氨酸。3個雜合性錯義突變中,c.5459T>C和點突變c.1190T>A為新發現突變位點。
先證者(Ⅱ2)父親(Ⅰ1)攜帶有c.1190T>A(p.I397K)突變,母親(Ⅰ2)攜帶有c.5459T>C(p.M1820T)和c.802G>A(p.G268R)2個突變位點。其余表型正常的家系成員和正常對照者均未同時檢測到這3個突變位點(圖6)。突變在該家系中呈現共分離狀態。
先證者(Ⅱ2)、先證者父親(Ⅰ1)、哥哥(Ⅱ1)、女兒(Ⅲ2)均攜帶另一個與USH有關的GPR98基因的雜合錯義突變c.4703G>A(p.S1568N)。先證者(Ⅱ2)、先證者母親(Ⅰ2)、哥哥(Ⅱ1)、兒子(Ⅲ1)均攜帶GPR98基因的雜合錯義突變c.15701A>G(p.K5234R)。突變在該家系中未出現共分離。
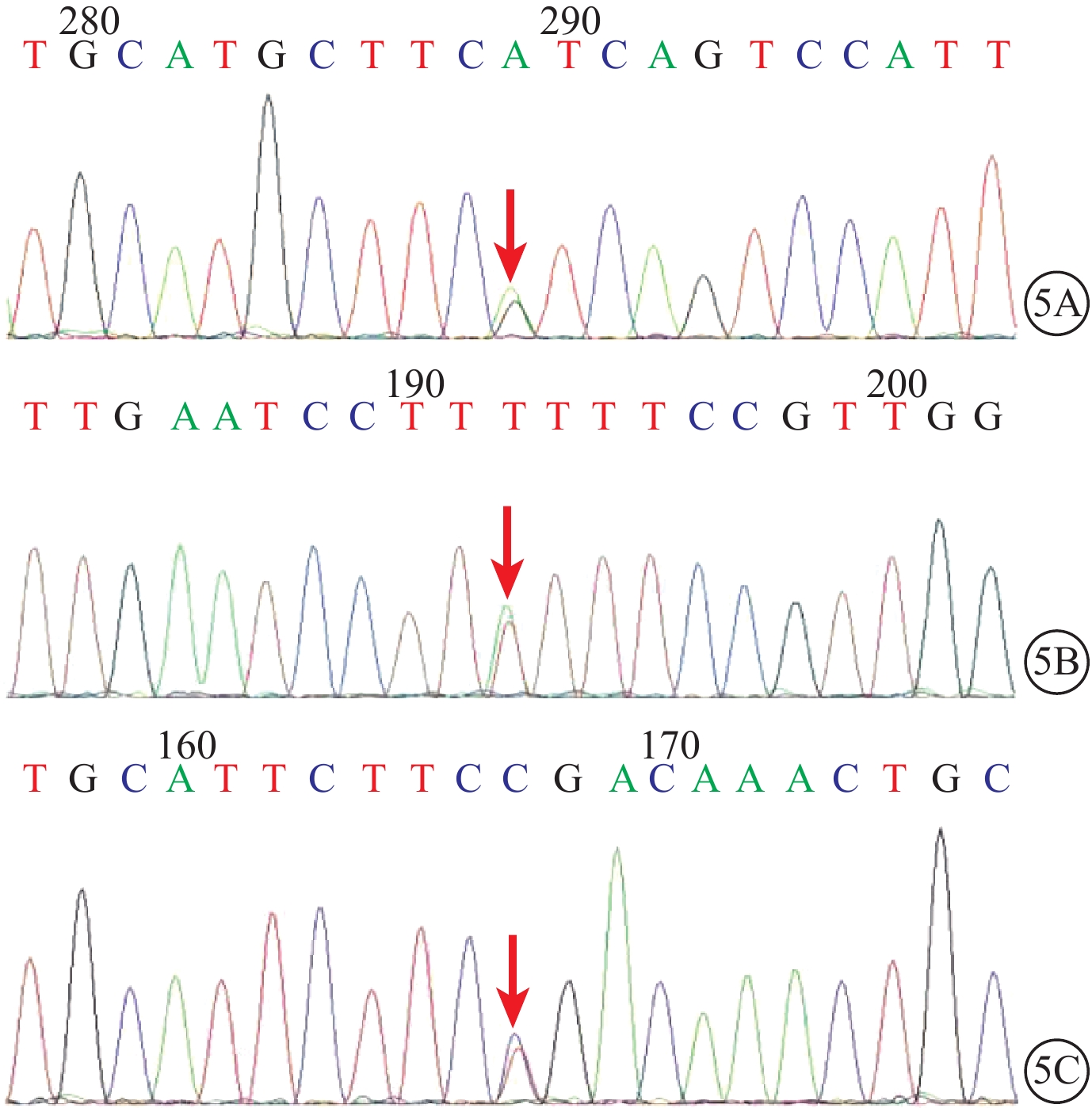
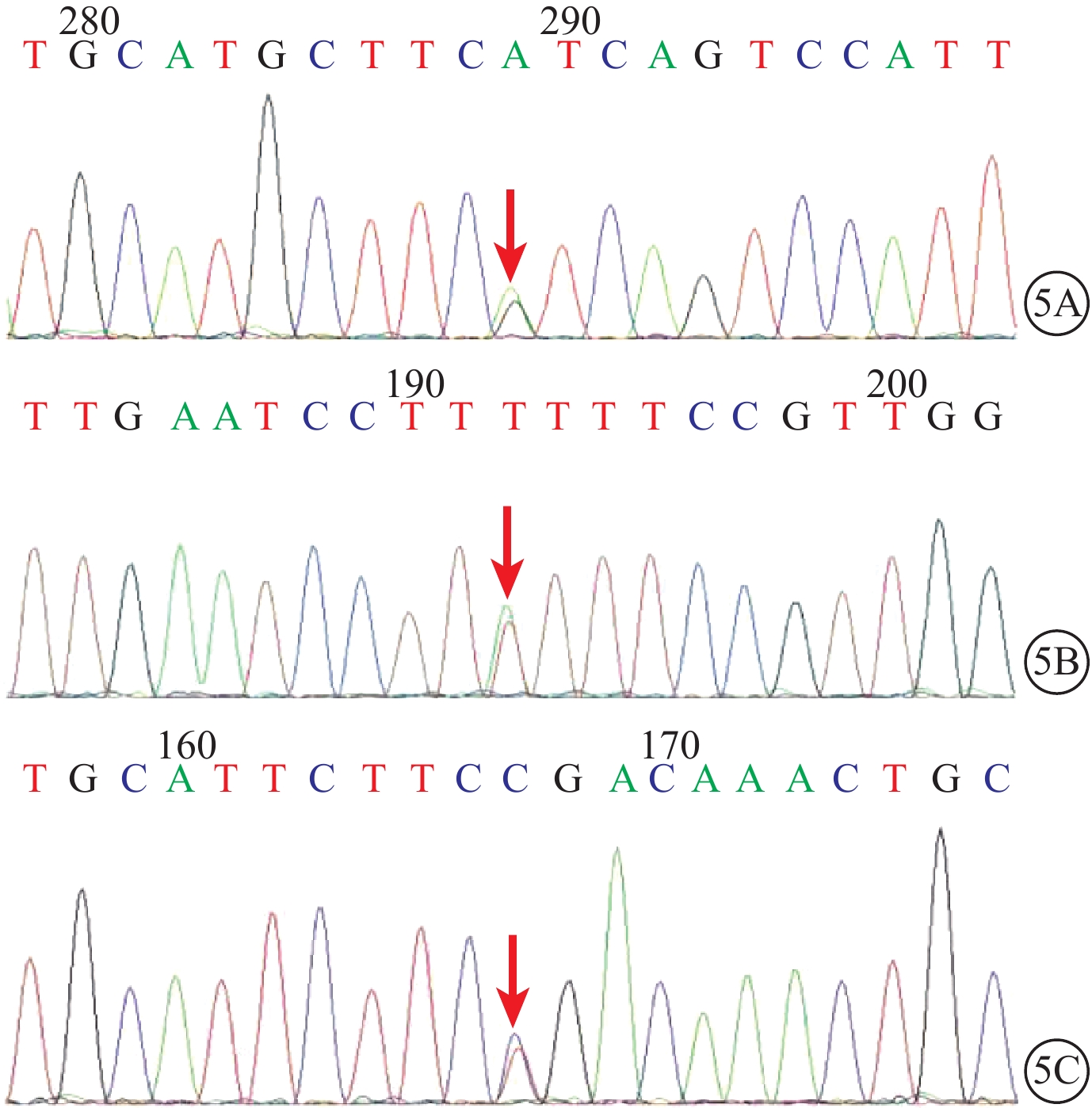 圖5
USH2患者USH基因測序圖。5A. USH2A基因第27號外顯子的c.5459T>C(p.M1820T)錯義突變(紅箭);5B. USH2A基因第7號外顯子的c.1190T>A(p.I397K)錯義突變(紅箭);5C. USH2A基因第5號外顯子的c.802G>A(p.G268R)錯義突變(紅箭)
圖5
USH2患者USH基因測序圖。5A. USH2A基因第27號外顯子的c.5459T>C(p.M1820T)錯義突變(紅箭);5B. USH2A基因第7號外顯子的c.1190T>A(p.I397K)錯義突變(紅箭);5C. USH2A基因第5號外顯子的c.802G>A(p.G268R)錯義突變(紅箭)
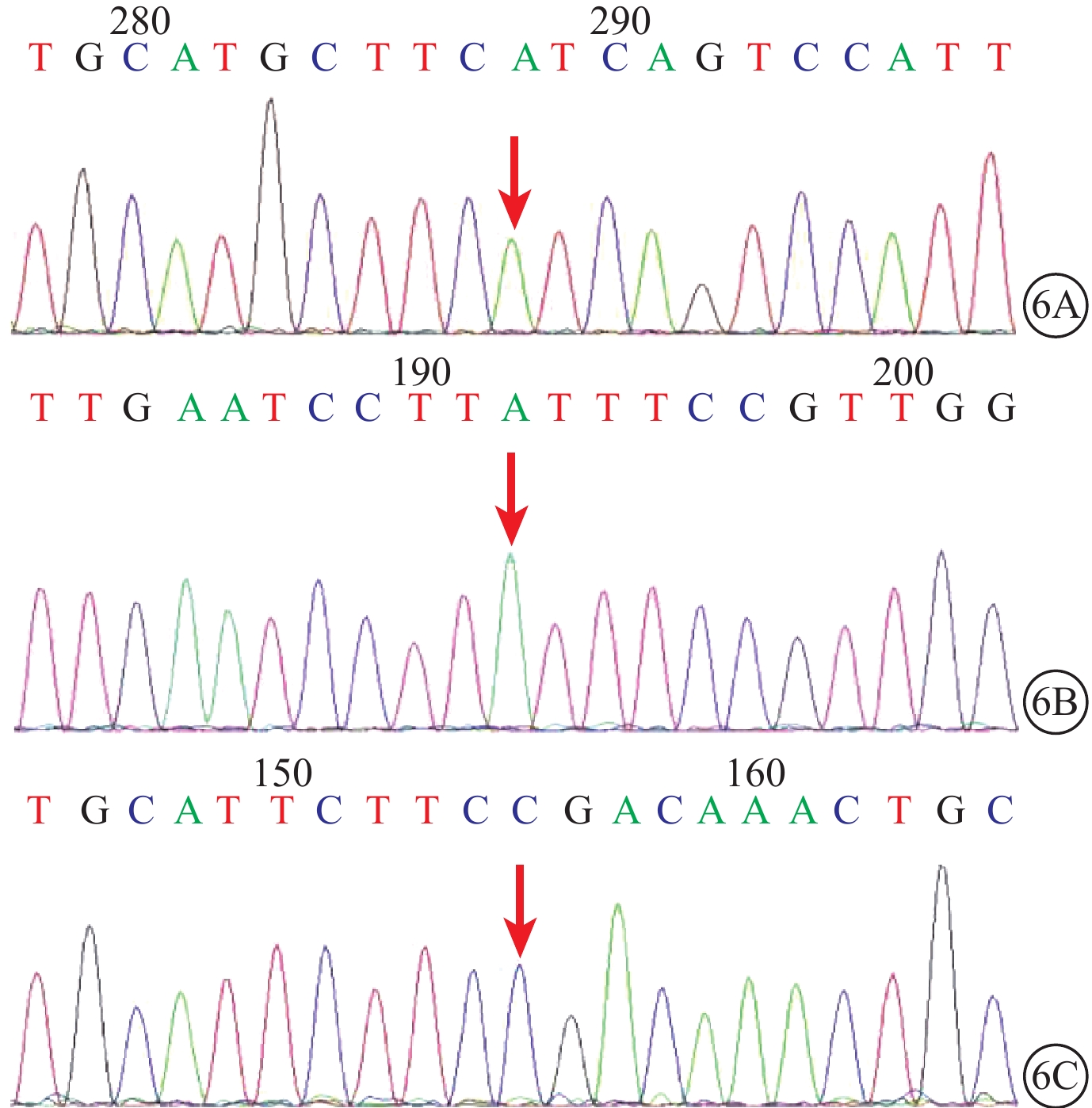
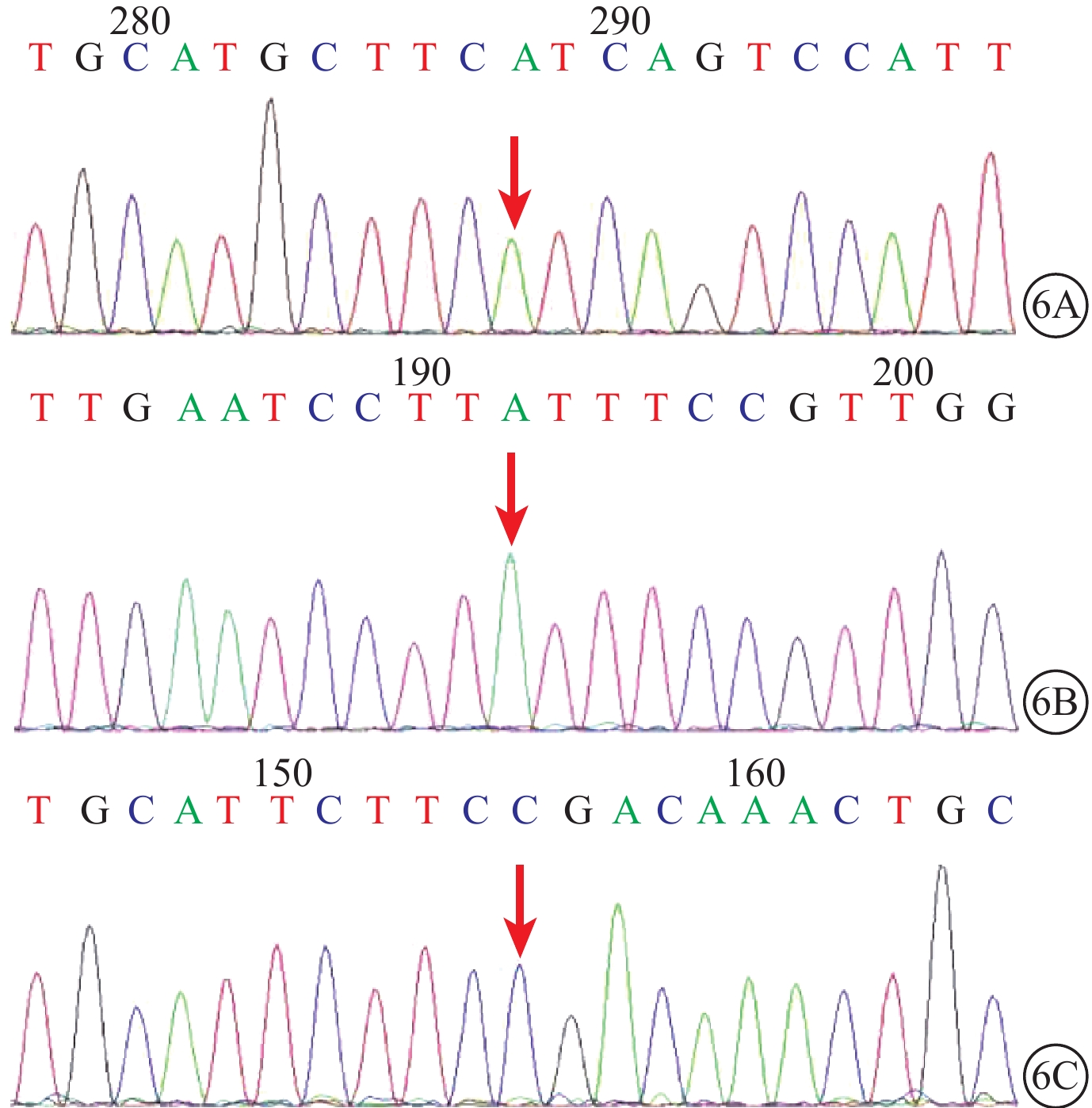 圖6
正常對照者USH基因測序圖。6A~6C. 分別為USH2A基因第27、7、5號外顯子外顯子5459、1190、802位點,均不存在突變
圖6
正常對照者USH基因測序圖。6A~6C. 分別為USH2A基因第27、7、5號外顯子外顯子5459、1190、802位點,均不存在突變
3 討論
迄今為止,已確定與USH相關的致病基因14個,其中MYO7A、USH1C、CDH23、PCDH15、SANS、CIB2等6個致病基因與USH1相關;USH2A、VLGR1、WHRN等3個致病基因與USH2相關[5, 8, 9];CLRN1與USH3相關[10]。此外HARS 和ABHD12也被確定與非典型USH3相關[11, 12]。2個修飾基因 PDZD7 和CEP250也被發現[13, 14]。USH2A是USH2最常見的致病基因[3]。因此,USH2A基因的測序篩查是診斷USH2的首選。
USH臨床特征和基因遺傳的高度異質性使得該疾病難以通過一代測序對其進行分子水平的診斷。因此,本研究采用目標區域捕獲聯合高通量測序技術對USH患者的致病基因突變進行檢測。結果發現USH2A基因的3個錯義突變c.5459T>C、c.1190T>A、c.802G>A,其中c.5459T>C、c.1190T>A為新發現的突變位點。已有文獻報道,錯義突變c.802G>A為USH2的相關致病突變[5]。本家系中攜帶c.802G>A和c.5459T>C2突變的受試者(Ⅰ2)表型正常,且無其他證據可以證明錯義突變c.802G>A為導致本家系發病的原因。因此,與既往報道錯義突變c.802G>A可能為致病突變不同[5],僅有c.802G>A點突變可能不足以導致USH2。疾病表型發生的具體機制尚不明確。因此,進行深入的分子生物學研究,了解USH2A基因突變對Usherin蛋白的結構和與其他蛋白間相互作用所造成的影響,以及闡明USH2A基因突變造成聽力損傷的病理生理機制十分必要。本家系中發現的3個雜合性錯義突變c.5459T>C、c.1190T>A、c.802G>A與疾病表型共分離,且在正常人數據庫對比中均未發現此3個突變同時出現。因此,我們考慮3個位點同時發生變異是導致本家系USH2的遺傳學基礎。先證者父親(Ⅰ1)攜帶c.1190T>A(p.I397K)突變,母親(Ⅰ2)攜帶c.5459T>C(p.M1820T)和c.802G>A(p.G268R)2個突變位點,根據基因檢測結果表明本家系的遺傳方式為常染色體隱性遺傳。
在GPR98基因上檢測到2個雜合性錯義突變c.4703G>A(p.S1568N)和c.15701A>G(p.K5234R),其中錯義突變c.4703G>A已有文獻報道與耳聾的發生相關[15]。但是本家系中僅攜帶該突變位點的家系成員(Ⅲ2)并未發生耳聾,所以我們認為該突變位點的致病性有待進一步確認。攜帶GPR98基因復合雜合突變(c.4703G>A和c.15701A>G)的家系成員(Ⅱ1)臨床表型正常,故考慮該復合雜合突變不是本家系的致病突變。先證者(Ⅱ2)同時攜帶GPR98的復合雜合突變和USH2A基因的3個雜合性錯義突變,患者(Ⅱ3)僅攜帶USH2A基因的3個雜合性錯義突變,而2例患者的臨床表型和臨床分型均一致,故推測GPR98基因的2個突變位點可能為非致病性突變,且可能對于USH2A基因的復合雜合突變的致病性無協同作用。
因此,本研究在USH2A基因上檢測到的3個雜合性錯義突變位點,c.5459T>C(p.M1820T)、c.1190T>A(p.I397K)和c.802G>A(p.G268R),構成復合雜合性性突變為本家系的致病原因。該家系成員在USH2A基因3個位點同時出現變異時才表現出臨床癥狀。
Usher綜合征(USH)是一種以耳聾和視網膜色素變性(RP)為特征的常染色體隱性遺傳疾病[1]。臨床上根據聽力和前庭功能受累情況將其分為1(USH1)、2(USH2)、3(USH3)等3種臨床亞型[2]。其中,USH2最為常見[3],表現為先天性中度-重度非漸進性耳聾,前庭功能正常,20歲左右出現RP癥狀[4, 5]。USH具有高度遺傳異質性,研究發現,超過70%的USH2是USH2A基因突變所致[6]。我們對一個USH2家系進行了相關致病基因檢測,旨在明確該家系患者的致病基因。現將結果報道如下。
1 對象和方法
本研究獲鄭州大學第一附屬醫院倫理委員會批準;嚴格遵守赫爾辛基宣言,所有受試者及未成年受試者監護人均簽署知情同意書。一個三代USH2家系(圖1)中2例患者及5名正常家系成員納入研究。所有受試者均行最佳矯正視力(BCVA)、裂隙燈顯微鏡、間接檢眼鏡、全視野視網膜電圖(ERG)、光相干斷層掃描(OCT)、視野檢查,以及前庭功能和純音測聽檢查。同時招募100名無血緣關系的健康志愿者作為正常對照者進行USH基因測序比對。
先證者(Ⅱ2),男,32歲。10歲時雙耳出現聽力下降;目前雙耳重度感音神經性耳聾,前庭功能未見異常。12歲時雙眼出現夜盲。雙眼BCVA分別為0.4、0.7。視盤顏色淡,視網膜血管變細,周邊彌漫性骨細胞樣色素顆粒沉著(圖2)。全視野ERG明適應、暗適應a、b波重度下降。OCT檢查可見黃斑中心凹以外區域視網膜明顯變薄,尤以視細胞層明顯(圖3)。視野向心性縮小(圖4)。先證者弟弟(Ⅱ3),30歲。雙耳重度感音神經性耳聾,前庭功能正常。雙眼BCVA分別為0.9、0.8。眼底表現及全視野ERG、OCT、視野檢查結果同先證者(Ⅱ2)。家系成員BCVA、眼底、視野、OCT、全視野ERG檢查均未見異常。依據臨床檢查結果并結合家系分析,患者符合常染色體隱性遺傳USH2診斷標準[7]。
圖1
患者家系圖。■:男性患者;□:正常男性;○:正常女性;↑:先證者;M1:p.I397K/c.1190T>A;M2:p.M182T/c.5459T>C;M3:G268R/c.802G>A
圖2
先證者(Ⅱ2)雙眼彩色眼底像。2A. 右眼;2B. 左眼。視盤顏色淡,視網膜血管變細;周邊部視網膜骨細胞樣色素沉著(白箭)
圖3
先證者(Ⅱ2)雙眼OCT像。3A. 右眼;3B. 左眼。黃斑中心凹以外區域視網膜明顯變薄,尤其以視細胞層更為明顯
圖4
先證者(Ⅱ2)視野檢查像。視野向心性縮小
采集家系中7名受試者的外周靜脈血3 ml,乙二胺四乙酸抗凝。采用天根生化科技(北京)有限公血液基因組DNA提取試劑盒按照標準操作流程提取全基因組DNA,紫外分光光度計定量。應用北京邁基諾公司提供的遺傳眼病目標基因試劑盒對目前已知的136個遺傳性視網膜疾病致病基因的外顯子區域進行捕獲。每例患者提取3 μg DNA作為起始量,采用超聲儀進行超聲片段化,打斷樣本片段為150堿基對,對其進行末端補平、兩端加A、連接adoptor、純化,文庫質量檢測后進行雜交、洗脫、連接介導聚合酶鏈反應(PCR)擴增對捕獲文庫富集。
PCR產物采用Beckman Ampure beads按1.8:1體積進行純化,應用II-lumina HiSeq 2500進行高通量測序。測序結果通過基因組數據庫、共識編碼序列計劃、NCBI數據庫單核苷酸多態性(SNP)子庫、單核苷酸多態性數據庫、千人基因組計劃、人基因組數據庫對SNP進行注釋,確定候選致病基因突變位點。
所有檢測出的可能致病突變均通過Sanger測序進行驗證并進行家系共分離驗證。Sanger測序結果再通過SIFT預測去掉不影響蛋白功能的SNPs,最終確定致病性基因突變位點。
2 結果
USH基因測序結果顯示,2例患者(Ⅱ2、Ⅱ3)的USH2A基因第27、7、5號外顯子分別存在c.5459T>C(p.M1820T)、c.1190T>A(p.I397K)、c.802G>A(p.G268R)3個雜合性錯義突變(圖5)。其中,27號外顯子第5 459位核糖核苷酸T變C,導致其編碼的第1820位氨基酸由甲硫氨酸變為蘇氨酸;7號外顯子第1190位核糖核苷酸T變A,導致其編碼的第397位氨基酸由異亮氨酸變為賴氨酸;5號外顯子第802位核糖核苷酸G變A,導致其編碼的第268位氨基酸由甘氨酸變為精氨酸。3個雜合性錯義突變中,c.5459T>C和點突變c.1190T>A為新發現突變位點。
先證者(Ⅱ2)父親(Ⅰ1)攜帶有c.1190T>A(p.I397K)突變,母親(Ⅰ2)攜帶有c.5459T>C(p.M1820T)和c.802G>A(p.G268R)2個突變位點。其余表型正常的家系成員和正常對照者均未同時檢測到這3個突變位點(圖6)。突變在該家系中呈現共分離狀態。
先證者(Ⅱ2)、先證者父親(Ⅰ1)、哥哥(Ⅱ1)、女兒(Ⅲ2)均攜帶另一個與USH有關的GPR98基因的雜合錯義突變c.4703G>A(p.S1568N)。先證者(Ⅱ2)、先證者母親(Ⅰ2)、哥哥(Ⅱ1)、兒子(Ⅲ1)均攜帶GPR98基因的雜合錯義突變c.15701A>G(p.K5234R)。突變在該家系中未出現共分離。
圖5
USH2患者USH基因測序圖。5A. USH2A基因第27號外顯子的c.5459T>C(p.M1820T)錯義突變(紅箭);5B. USH2A基因第7號外顯子的c.1190T>A(p.I397K)錯義突變(紅箭);5C. USH2A基因第5號外顯子的c.802G>A(p.G268R)錯義突變(紅箭)
圖6
正常對照者USH基因測序圖。6A~6C. 分別為USH2A基因第27、7、5號外顯子外顯子5459、1190、802位點,均不存在突變
3 討論
迄今為止,已確定與USH相關的致病基因14個,其中MYO7A、USH1C、CDH23、PCDH15、SANS、CIB2等6個致病基因與USH1相關;USH2A、VLGR1、WHRN等3個致病基因與USH2相關[5, 8, 9];CLRN1與USH3相關[10]。此外HARS 和ABHD12也被確定與非典型USH3相關[11, 12]。2個修飾基因 PDZD7 和CEP250也被發現[13, 14]。USH2A是USH2最常見的致病基因[3]。因此,USH2A基因的測序篩查是診斷USH2的首選。
USH臨床特征和基因遺傳的高度異質性使得該疾病難以通過一代測序對其進行分子水平的診斷。因此,本研究采用目標區域捕獲聯合高通量測序技術對USH患者的致病基因突變進行檢測。結果發現USH2A基因的3個錯義突變c.5459T>C、c.1190T>A、c.802G>A,其中c.5459T>C、c.1190T>A為新發現的突變位點。已有文獻報道,錯義突變c.802G>A為USH2的相關致病突變[5]。本家系中攜帶c.802G>A和c.5459T>C2突變的受試者(Ⅰ2)表型正常,且無其他證據可以證明錯義突變c.802G>A為導致本家系發病的原因。因此,與既往報道錯義突變c.802G>A可能為致病突變不同[5],僅有c.802G>A點突變可能不足以導致USH2。疾病表型發生的具體機制尚不明確。因此,進行深入的分子生物學研究,了解USH2A基因突變對Usherin蛋白的結構和與其他蛋白間相互作用所造成的影響,以及闡明USH2A基因突變造成聽力損傷的病理生理機制十分必要。本家系中發現的3個雜合性錯義突變c.5459T>C、c.1190T>A、c.802G>A與疾病表型共分離,且在正常人數據庫對比中均未發現此3個突變同時出現。因此,我們考慮3個位點同時發生變異是導致本家系USH2的遺傳學基礎。先證者父親(Ⅰ1)攜帶c.1190T>A(p.I397K)突變,母親(Ⅰ2)攜帶c.5459T>C(p.M1820T)和c.802G>A(p.G268R)2個突變位點,根據基因檢測結果表明本家系的遺傳方式為常染色體隱性遺傳。
在GPR98基因上檢測到2個雜合性錯義突變c.4703G>A(p.S1568N)和c.15701A>G(p.K5234R),其中錯義突變c.4703G>A已有文獻報道與耳聾的發生相關[15]。但是本家系中僅攜帶該突變位點的家系成員(Ⅲ2)并未發生耳聾,所以我們認為該突變位點的致病性有待進一步確認。攜帶GPR98基因復合雜合突變(c.4703G>A和c.15701A>G)的家系成員(Ⅱ1)臨床表型正常,故考慮該復合雜合突變不是本家系的致病突變。先證者(Ⅱ2)同時攜帶GPR98的復合雜合突變和USH2A基因的3個雜合性錯義突變,患者(Ⅱ3)僅攜帶USH2A基因的3個雜合性錯義突變,而2例患者的臨床表型和臨床分型均一致,故推測GPR98基因的2個突變位點可能為非致病性突變,且可能對于USH2A基因的復合雜合突變的致病性無協同作用。
因此,本研究在USH2A基因上檢測到的3個雜合性錯義突變位點,c.5459T>C(p.M1820T)、c.1190T>A(p.I397K)和c.802G>A(p.G268R),構成復合雜合性性突變為本家系的致病原因。該家系成員在USH2A基因3個位點同時出現變異時才表現出臨床癥狀。