引用本文: 鄭衛東, 黃焱, 萬松, 徐國興. 糖尿病大鼠后期胰島素強化治療對視網膜中谷氨酸與γ-氨基丁酸表達的影響. 中華眼底病雜志, 2015, 31(6): 586-590. doi: 10.3760/cma.j.issn.1005-1015.2015.06.018 復制
高糖“代謝記憶”是指糖尿病(DM)患者的高血糖水平在早期如果不能得到控制,即使后期血糖水平持續穩定在正常水平,糖尿病視網膜病變(DR)等DM慢性并發癥仍然持續進展[1]。近年相關研究發現,細胞凋亡等多種機制參與了DR相關“代謝記憶”的發生[2]。有研究發現,后期胰島素強化治療不能抑制初期高血糖導致凋亡因子的持續上調[3]。現有研究認為,DR也屬于神經退行性病變,神經組織病變早于視網膜血管改變[4, 5],視網膜神經細胞凋亡是其基本病理過程。谷氨酸(Glu)是視網膜中最主要的興奮性神經遞質,γ-氨基丁酸(GABA)為重要的抑制神經遞質,能拮抗Glu的興奮毒性,通常兩者之間保持平衡。但在視網膜缺血時,Glu釋放增多,誘導GABA釋放增加但其升高幅度不及Glu,兩者紊亂表達產生的Glu興奮毒性導致視網膜神經細胞凋亡[6]。為此,我們通過后期胰島素強化治療,觀察DM大鼠視網膜組織中Glu、GABA表達和神經細胞凋亡情況,初步探討高糖“代謝記憶”致DM大鼠視網膜神經病變的機制。現將結果報道如下。
1 材料和方法
鏈脲佐菌素(STZ,美國Sigma公司);精蛋白生物合成人胰島素(丹麥諾和諾德公司);即用型Biotin SP-HRP免疫組織化學試劑盒(北京鼎國生物技術有限公司);原位末端標記法(TUNEL)細胞凋亡檢測試劑盒(北京中杉金橋生物技術有限公司);光學顯微鏡(BX51,日本Olympus公司);顯微照相系統(PM-20,日本Olympus公司);高效液相色譜儀(美國Agilent公司);色譜柱(XDB-C18,美國Agilent公司);血糖儀(德國羅氏公司)。
雄性Sprague Dawley大鼠60只,6周齡,體重180~220 g,清潔級,上海斯萊克實驗動物有限公司提供(許可證號:SCXK-2007-0005)。清潔實驗室飼養,自由攝食、飲水。應用隨機數字表法,將大鼠隨機分為DM組和正常對照組(NC組),分別為45、15只大鼠。DM組大鼠按65 mg/kg劑量一次腹腔注射1%STZ制作DM模型;建模后72 h尾靜脈采血測定空腹血糖≥16.7 mmol/L視為建模成功[7]。NC組大鼠腹腔注射檸檬酸-檸檬酸鈉緩沖液0.1 mmol/L。建模后,DM組隨機分為DM1組、DM2組、代謝記憶組(MM組),每組均為15只大鼠。6周時,MM組大鼠給予胰島素強化治療,早晚皮下注射精蛋白生物合成人胰島素,單只大鼠總用量6~8 IU/d,連續6周;治療期間維持血糖值≤8.3 mmol/L[3]。DM1組大鼠建模后6周,MM組、DM2組、NC組建模后12周過量麻醉處死所有大鼠,手術顯微鏡分離雙眼視網膜組織。
高效液相色譜法(HPLC)[8]檢測大鼠視網膜組織中Glu、GABA的含量。視網膜勻漿,離心半徑6 cm,12 000 r/min離心20 min,取上清液,于-70℃液氮中保存。按制作的標準曲線,計算樣品中Glu、GABA的濃度C,按含量=C×V×f/W(nmol/g)(C:對照品溶液濃度,V:無水乙醇體積,W:取樣量,f:換算因子),計算視網膜中Glu、GABA含量。
實時定量熒光聚合酶鏈反應(RT-PCR)檢測視網膜中谷氨酸脫羧酶(GAD)mRNA的表達。按照Trizol試劑盒說明書提取視網膜總RNA。3-磷酸甘油醛脫氫酶(GAPDH)為內參照基因,設計針對大鼠GAD和β-肌動蛋白(β-actin)的特異性引物。GAD上游引物:5′-CCTGCTAGAGCTGTTCCCAC-3′,下游引物:5′-CCCTGCCCAAAGATAGACAA-3′,擴增產物為199堿基對(bp)。β-actin為內參照,上游引物:5′-ACAGCAACAGGGTGGTGGAC-3′,下游引物:5′-TTTGAGGGTGCAGCGAACTT-3′,擴增產物為252 bp。反應條件:94℃預變性2 min,94℃變性30 s、56℃退火30 s、72℃延伸30 s、35循環,72℃延伸10 min。瓊脂糖凝膠電泳,凝膠成像系統觀察聚合酶鏈反應產物的電泳帶。圖像分析軟件one-Dscan分析目標條帶的灰度值,各組以目的基因(GAD)的吸光度[A,舊稱光密度(OD)]值與內參照基因(GADPH)A值的比值表示目的基因相對表達量。
TUNEL法檢測視網膜神經細胞凋亡。眼球摘除后置于10%中性福爾馬林中固定24 h,常規脫水透明,石蠟包埋,作4μm厚度的連續切片。按試劑盒說明進行操作,細胞核呈棕黃或棕褐色為凋亡陽性細胞。光學顯微鏡下觀察視網膜神經細胞染色情況,統計細胞凋亡率并計算凋亡指數。凋亡指數=陽性細胞數/同一視野細胞總數×100%。每個標本取3張切片,每張切片在400倍視野下計數5個不同視野,取平均值作為該標本的凋亡指數。
SPSS 19.0統計學軟件進行統計學分析處理。數據以均數±標準差(
2 結果
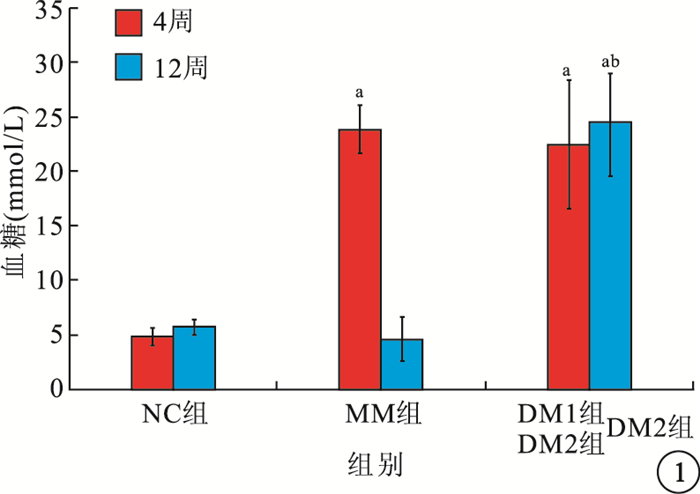
造模后4周,MM組、DM1、DM2組大鼠空腹血糖明顯高于NC組,差異有統計學意義(F=97.916,P<0.01);MM組大鼠空腹血糖與DM組比較,差異無統計學意義(t=0.891,P>0.05)。6周,MM組大鼠經胰島素強化治療后,大鼠空腹血糖降低至正常水平。12周,MM組、NC組大鼠空腹血糖均低于DM2組,差異有統計學意義(F=225.316,P<0.01),且MM組與NC組之間無顯著差異(t=1.989,P>0.05)(圖 1)。
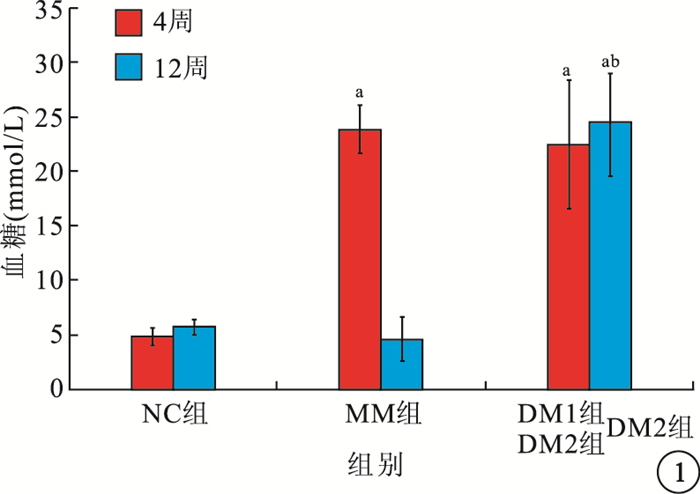 圖1
各組大鼠4周及DM2組、MM組、NC組大鼠12周血糖值比較。a與NC組比較,P<0.05;b與MM組比較,P<0.05
圖1
各組大鼠4周及DM2組、MM組、NC組大鼠12周血糖值比較。a與NC組比較,P<0.05;b與MM組比較,P<0.05
HPLC檢測結果顯示,MM組、DM1組、DM2組大鼠視網膜組織中Glu(F=104.108)、GABA(F=35.088)含量及其Glu/GABA比值(F=57.436)均較NC組高,差異有統計學意義(P<0.05);MM組大鼠視網膜中Glu(t=6.963)、GABA(t=4.385)含量及其Glu/GABA比值(t=4.163)較DM1組高,差異有統計學意義(P<0.05);MM組大鼠視網膜中Glu(t=3.411)、GABA(t=3.709)含量較DM2組低,差異有統計學意義(P<0.05),Glu/GABA比值與DM2組比較,差異無統計學意義(t=1.199,P>0.05)(表 1)。
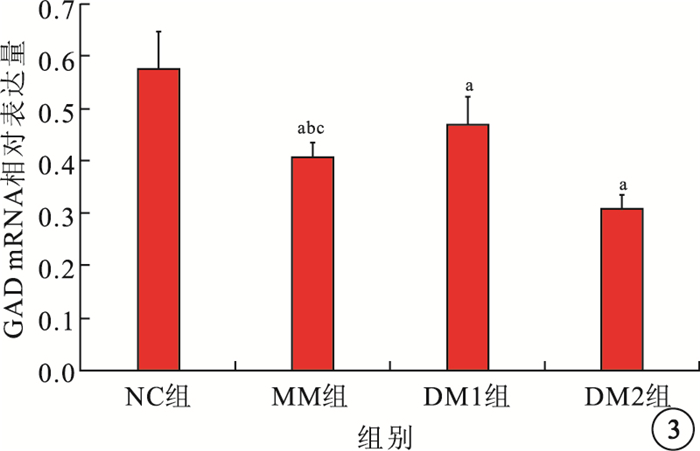
RT-PCR檢測結果顯示,NC組、DM1組、MM組、DM2組大鼠視網膜組織中GAD mRNA相對表達量分別為0.577±0.069、0.472±0.051、0.409±0.027、0.311±0.024。MM組、DM1組、DM2組大鼠視網膜中GAD mRNA相對表達量均較NC組低,差異有統計學意義(F=57.668,P<0.05);MM組大鼠視網膜中GAD mRNA相對表達量較DM1組低(t=3.496, P<0.05),較DM2組高(t=8.613, P<0.05),差異有統計學意義(圖 2, 3)。
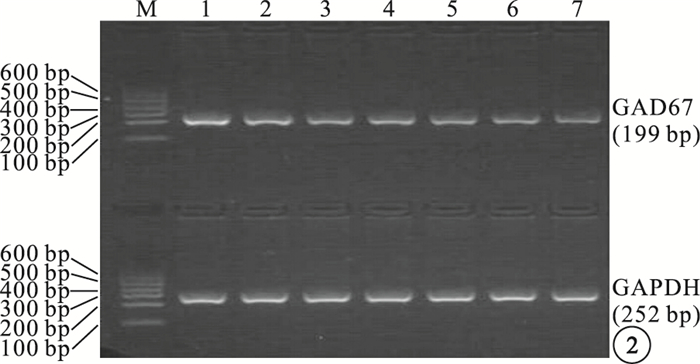
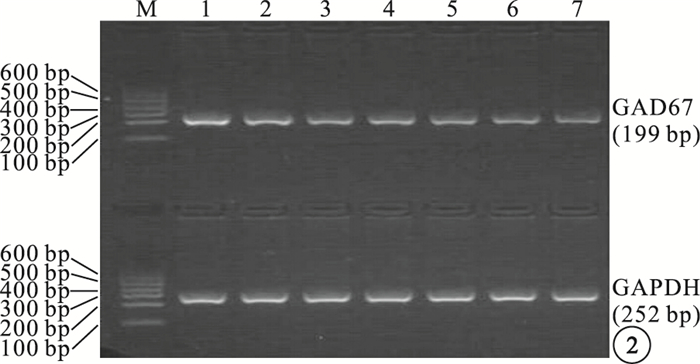 圖2
各組細胞大鼠視網膜組織中GAD mRNA相對表達量電泳圖。M:Marker;1:NC組;2~3:DM1組;4~5:MM組;6~7:DM2組
圖2
各組細胞大鼠視網膜組織中GAD mRNA相對表達量電泳圖。M:Marker;1:NC組;2~3:DM1組;4~5:MM組;6~7:DM2組
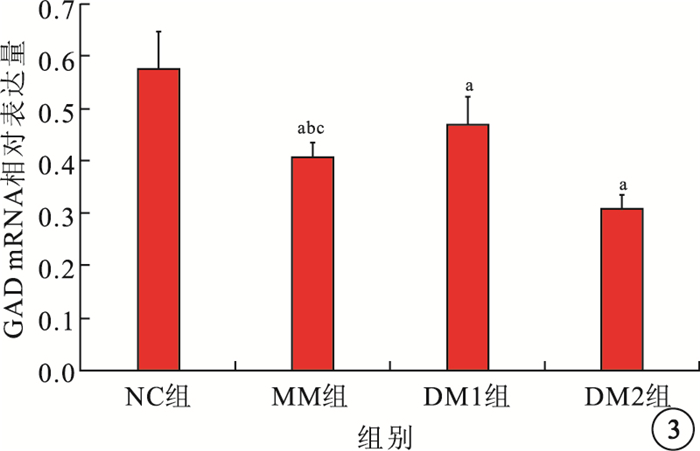 圖3
各組大鼠視網膜組織中GAD mRNA相對表達量比較。a與NC組比較,P<0.05;b與DM1組比較,P<0.05;c與DM2組比較,P<0.05
圖3
各組大鼠視網膜組織中GAD mRNA相對表達量比較。a與NC組比較,P<0.05;b與DM1組比較,P<0.05;c與DM2組比較,P<0.05
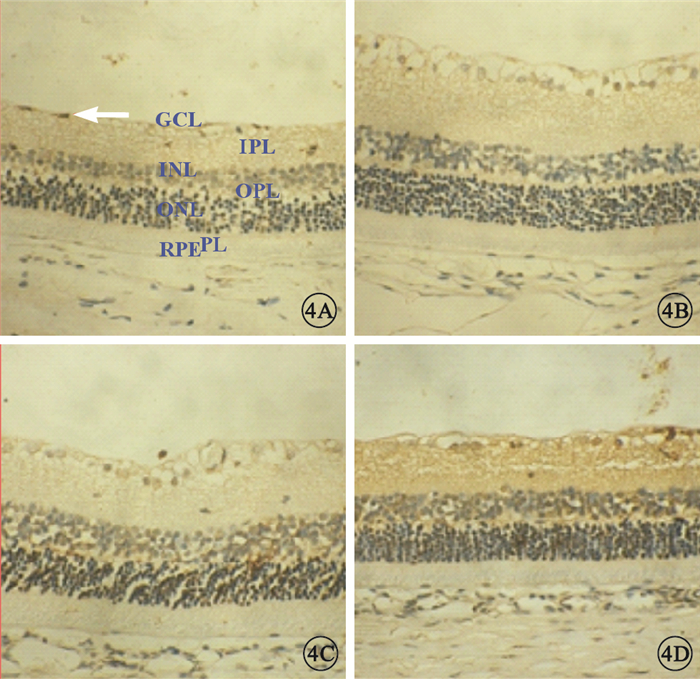
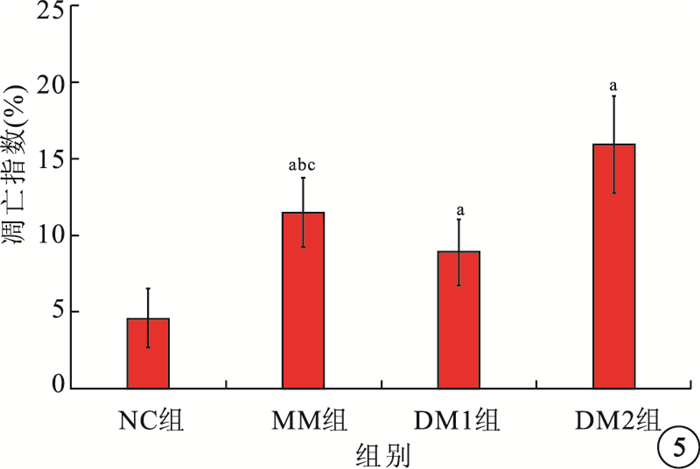
TUNEL法檢測結果顯示,NC組大鼠視網膜組織中凋亡細胞細胞核呈棕褐色,點狀分布于神經節細胞層(GCL)、內核層(INL);MM組、DM1組、DM2組大鼠視網膜中凋亡細胞顯著增多,DM2組較DM1組增多更明顯。MM組大鼠視網膜中凋亡細胞較DM2組減少,但較NC組和DM1組仍增多(圖 4)。MM組、DM1組、DM2組凋亡指數均較NC組高,差異有統計學意義(F=37.385,P<0.05);MM組凋亡指數較DM1組高(t=2.584, P<0.05),較DM2組低(t=3.531, P<0.05),差異有統計學意義(圖 5)。
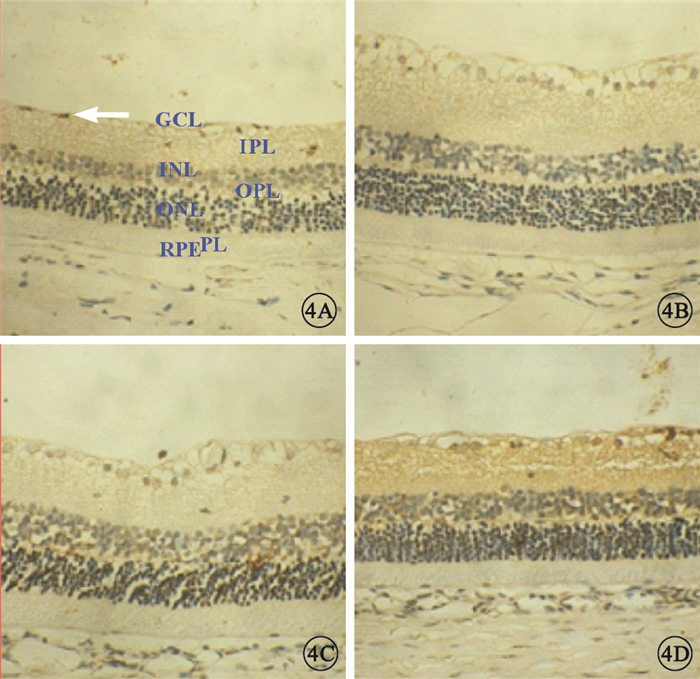 圖4
各組大鼠視網膜組織光學顯微鏡像。4A.NC組,視網膜中僅見點狀陽性細胞(白箭);4B.DM1組,視網膜GCL、INL陽性細胞數量較NC組增多、密度增大;4C.MM組,視網膜GCL、INL陽性細胞數量較DM1組增多;4D.DM2組,視網膜可見大量陽性細胞。IPL:內叢狀層,OPL:外叢狀層,ONL:外核層,PL:視錐視桿細胞層,RPE:視網膜色素上皮二氨基聯苯胺×400
圖4
各組大鼠視網膜組織光學顯微鏡像。4A.NC組,視網膜中僅見點狀陽性細胞(白箭);4B.DM1組,視網膜GCL、INL陽性細胞數量較NC組增多、密度增大;4C.MM組,視網膜GCL、INL陽性細胞數量較DM1組增多;4D.DM2組,視網膜可見大量陽性細胞。IPL:內叢狀層,OPL:外叢狀層,ONL:外核層,PL:視錐視桿細胞層,RPE:視網膜色素上皮二氨基聯苯胺×400
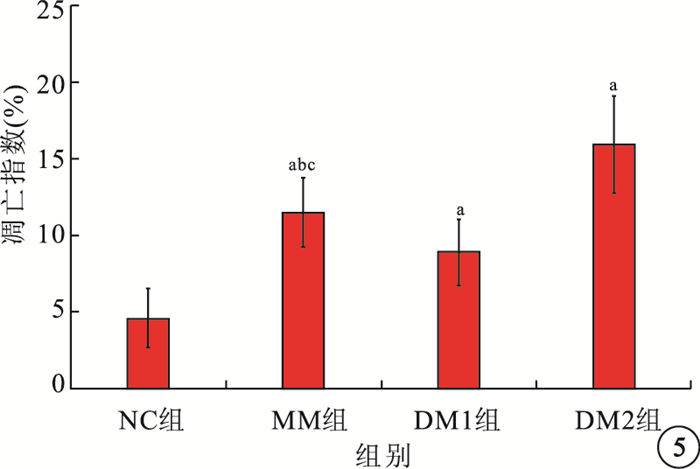 圖5
各組大鼠視網膜神經細胞凋亡指數比較。a與NC組比較,P<0.05;b與DM1組比較,P<0.05;c與DM2組比較,P<0.05
圖5
各組大鼠視網膜神經細胞凋亡指數比較。a與NC組比較,P<0.05;b與DM1組比較,P<0.05;c與DM2組比較,P<0.05
3 討論
長期慢性高血糖可導致DR發生發展,然而,DR等并發癥并不能完全由后期單純控制血糖來阻止。DM控制及并發癥試驗組臨床試驗、DM干預和并發癥流行病學調查試驗[9]和Holman等[10]經過長期隨訪發現,DM患者控制血糖后短期內并不能延緩DM并發癥的進展,即血管內皮細胞似乎對早期血糖環境具有記憶能力。目前, 高糖“代謝記憶”發生機制的研究是一個熱點, 但是多數研究都是圍繞高糖“記憶”損害視網膜微血管的機制展開,對視網膜神經病變的代謝記憶研究尚少。本研究重在揭示神經遞質紊亂和神經細胞凋亡在“代謝記憶”影響DR中的作用。
STZ誘導的DM鼠, 4~5周高血糖時發現星形膠質細胞激活以及數量增加[11, 12],同時也證明在膠質細胞神經膠質纖維酸性蛋白上調和反應性膠質增生[12]。從6周開始視網膜神經節細胞(RGC)數量減少[13],高血糖6周時可以鑒定RGC和血管細胞的細胞凋亡[14]。本課題組前期預實驗發現,高血糖4周時視神經細胞病變尚不明顯,8周或12周開始胰島素治療時血糖已很難控制。故本研究以6周為視網膜視神經病變研究起點。根據Bixler等[15]方法,在DM病程6周后MM組大鼠采用胰島素強化治療,結果顯示其前6周血糖與DM組大鼠血糖均處于高水平,治療后血糖下降并維持正常水平至12周。
Glu是視網膜中最主要的興奮性神經遞質,高血糖會引起視網膜缺血缺氧,引起Glu釋放增多,同時由于缺乏三磷酸腺苷,Müller細胞清除Glu能力減弱,最終導致突觸間隙中Glu堆積并激活Glu受體,Ca2+過度內流、超載,啟動系列級聯反應引起細胞離子滲透壓和電化學性質改變,產生興奮毒性,導致神經細胞凋亡。Gu等[16]認為Glu升高可誘導半胱氨酸蛋白酶非依賴的凋亡途徑,是DM視網膜神經細胞凋亡的原因之一。相關研究發現,DM大鼠視網膜中Glu含量的升高,導致血管內皮生長因子分泌增加,視網膜新生血管增多[17, 18]。由于Glu在DR發病中具有突出的作用, 本研究在DM大鼠12周后各組大鼠視網膜行HPLC檢測,MM組和DM2組大鼠視網膜組織中Glu含量相比NC組明顯升高,且視網膜神經細胞凋亡數也明顯增多。MM組在胰島素強化治療后相比DM2組Glu含量和視網膜神經細胞凋亡數降低,但仍未恢復至正常水平,說明MM組存在高糖“代謝記憶”,血糖雖然降至正常,Glu和細胞凋亡的增加仍然存在,DR持續進展。
GABA是一種重要的抑制性神經遞質,能拮抗Glu的興奮毒性,具有中樞和視網膜抑制保護作用,通常兩者之間保持平衡,Glu/GABA比值穩定。Glu被Müller細胞膜上的谷氨酸轉運體迅速轉運入胞質,既可以由谷氨酰胺合成酶轉化為無毒的谷氨酰胺,也可以經限速酶GAD催化生成GABA。視網膜缺血時,Glu釋放增多,誘導GABA釋放增加。本研究結果也顯示,MM組、DM2組大鼠視網膜組織中GABA含量相比NC組明顯升高,MM組在胰島素強化治療后相比DM2組GABA含量部分降低,但仍未恢復至正常水平。Glu/GABA比值相比Glu含量更能反應氨基酸的興奮毒性和神經功能的異常改變。本課題組前期研究證實,在DM早期大鼠視網膜組織中GABA升高,但其升高幅度不及Glu,Glu/GABA比值仍顯著升高,氨基酸毒性超過了機體的保護作用,視網膜缺血損傷繼續發生[8]。對于Glu/GABA比值,本研究發現MM組、DM2組大鼠視網膜組織中Glu/GABA的比值相比NC組均明顯升高,MM組在胰島素強化治療后相比DM2組其比值并無明顯改變,說明胰島素強化治療對前期高血糖引起的Glu/GABA比值升高無效。GAD是合成GABA的限速酶,在Müller細胞中表達豐富,GABA合成過多可抑制GAD的表達,形成負反饋。本研究結果也證實了這點,MM組、DM2組大鼠視網膜組織中GAD表達相比NC組均明顯降低,MM組在胰島素強化治療后相比DM2組GAD表達部分升高,但仍未恢復至正常水平。
前期高血糖引起的神經遞質紊亂,經后期胰島素強化治療,只能獲得部分改善,甚至對于升高的Glu/GABA比值無任何影響,視網膜神經組織仍存在Glu過興奮毒性,視網膜神經病變繼續發生發展。Glu、GABA紊亂可能是高糖“代謝記憶”致DR的機制之一。本研究提出了高糖“代謝記憶”機制的新思路,但尚未明確遞質紊亂與高糖“記憶”引起的視網膜病變存在必然的因果關系,后續將進一步研究如何降低興奮性遞質的含量,其改變是否能改善高糖“記憶”對視網膜的損害。
高糖“代謝記憶”是指糖尿病(DM)患者的高血糖水平在早期如果不能得到控制,即使后期血糖水平持續穩定在正常水平,糖尿病視網膜病變(DR)等DM慢性并發癥仍然持續進展[1]。近年相關研究發現,細胞凋亡等多種機制參與了DR相關“代謝記憶”的發生[2]。有研究發現,后期胰島素強化治療不能抑制初期高血糖導致凋亡因子的持續上調[3]。現有研究認為,DR也屬于神經退行性病變,神經組織病變早于視網膜血管改變[4, 5],視網膜神經細胞凋亡是其基本病理過程。谷氨酸(Glu)是視網膜中最主要的興奮性神經遞質,γ-氨基丁酸(GABA)為重要的抑制神經遞質,能拮抗Glu的興奮毒性,通常兩者之間保持平衡。但在視網膜缺血時,Glu釋放增多,誘導GABA釋放增加但其升高幅度不及Glu,兩者紊亂表達產生的Glu興奮毒性導致視網膜神經細胞凋亡[6]。為此,我們通過后期胰島素強化治療,觀察DM大鼠視網膜組織中Glu、GABA表達和神經細胞凋亡情況,初步探討高糖“代謝記憶”致DM大鼠視網膜神經病變的機制。現將結果報道如下。
1 材料和方法
鏈脲佐菌素(STZ,美國Sigma公司);精蛋白生物合成人胰島素(丹麥諾和諾德公司);即用型Biotin SP-HRP免疫組織化學試劑盒(北京鼎國生物技術有限公司);原位末端標記法(TUNEL)細胞凋亡檢測試劑盒(北京中杉金橋生物技術有限公司);光學顯微鏡(BX51,日本Olympus公司);顯微照相系統(PM-20,日本Olympus公司);高效液相色譜儀(美國Agilent公司);色譜柱(XDB-C18,美國Agilent公司);血糖儀(德國羅氏公司)。
雄性Sprague Dawley大鼠60只,6周齡,體重180~220 g,清潔級,上海斯萊克實驗動物有限公司提供(許可證號:SCXK-2007-0005)。清潔實驗室飼養,自由攝食、飲水。應用隨機數字表法,將大鼠隨機分為DM組和正常對照組(NC組),分別為45、15只大鼠。DM組大鼠按65 mg/kg劑量一次腹腔注射1%STZ制作DM模型;建模后72 h尾靜脈采血測定空腹血糖≥16.7 mmol/L視為建模成功[7]。NC組大鼠腹腔注射檸檬酸-檸檬酸鈉緩沖液0.1 mmol/L。建模后,DM組隨機分為DM1組、DM2組、代謝記憶組(MM組),每組均為15只大鼠。6周時,MM組大鼠給予胰島素強化治療,早晚皮下注射精蛋白生物合成人胰島素,單只大鼠總用量6~8 IU/d,連續6周;治療期間維持血糖值≤8.3 mmol/L[3]。DM1組大鼠建模后6周,MM組、DM2組、NC組建模后12周過量麻醉處死所有大鼠,手術顯微鏡分離雙眼視網膜組織。
高效液相色譜法(HPLC)[8]檢測大鼠視網膜組織中Glu、GABA的含量。視網膜勻漿,離心半徑6 cm,12 000 r/min離心20 min,取上清液,于-70℃液氮中保存。按制作的標準曲線,計算樣品中Glu、GABA的濃度C,按含量=C×V×f/W(nmol/g)(C:對照品溶液濃度,V:無水乙醇體積,W:取樣量,f:換算因子),計算視網膜中Glu、GABA含量。
實時定量熒光聚合酶鏈反應(RT-PCR)檢測視網膜中谷氨酸脫羧酶(GAD)mRNA的表達。按照Trizol試劑盒說明書提取視網膜總RNA。3-磷酸甘油醛脫氫酶(GAPDH)為內參照基因,設計針對大鼠GAD和β-肌動蛋白(β-actin)的特異性引物。GAD上游引物:5′-CCTGCTAGAGCTGTTCCCAC-3′,下游引物:5′-CCCTGCCCAAAGATAGACAA-3′,擴增產物為199堿基對(bp)。β-actin為內參照,上游引物:5′-ACAGCAACAGGGTGGTGGAC-3′,下游引物:5′-TTTGAGGGTGCAGCGAACTT-3′,擴增產物為252 bp。反應條件:94℃預變性2 min,94℃變性30 s、56℃退火30 s、72℃延伸30 s、35循環,72℃延伸10 min。瓊脂糖凝膠電泳,凝膠成像系統觀察聚合酶鏈反應產物的電泳帶。圖像分析軟件one-Dscan分析目標條帶的灰度值,各組以目的基因(GAD)的吸光度[A,舊稱光密度(OD)]值與內參照基因(GADPH)A值的比值表示目的基因相對表達量。
TUNEL法檢測視網膜神經細胞凋亡。眼球摘除后置于10%中性福爾馬林中固定24 h,常規脫水透明,石蠟包埋,作4μm厚度的連續切片。按試劑盒說明進行操作,細胞核呈棕黃或棕褐色為凋亡陽性細胞。光學顯微鏡下觀察視網膜神經細胞染色情況,統計細胞凋亡率并計算凋亡指數。凋亡指數=陽性細胞數/同一視野細胞總數×100%。每個標本取3張切片,每張切片在400倍視野下計數5個不同視野,取平均值作為該標本的凋亡指數。
SPSS 19.0統計學軟件進行統計學分析處理。數據以均數±標準差(
2 結果
造模后4周,MM組、DM1、DM2組大鼠空腹血糖明顯高于NC組,差異有統計學意義(F=97.916,P<0.01);MM組大鼠空腹血糖與DM組比較,差異無統計學意義(t=0.891,P>0.05)。6周,MM組大鼠經胰島素強化治療后,大鼠空腹血糖降低至正常水平。12周,MM組、NC組大鼠空腹血糖均低于DM2組,差異有統計學意義(F=225.316,P<0.01),且MM組與NC組之間無顯著差異(t=1.989,P>0.05)(圖 1)。
圖1
各組大鼠4周及DM2組、MM組、NC組大鼠12周血糖值比較。a與NC組比較,P<0.05;b與MM組比較,P<0.05
HPLC檢測結果顯示,MM組、DM1組、DM2組大鼠視網膜組織中Glu(F=104.108)、GABA(F=35.088)含量及其Glu/GABA比值(F=57.436)均較NC組高,差異有統計學意義(P<0.05);MM組大鼠視網膜中Glu(t=6.963)、GABA(t=4.385)含量及其Glu/GABA比值(t=4.163)較DM1組高,差異有統計學意義(P<0.05);MM組大鼠視網膜中Glu(t=3.411)、GABA(t=3.709)含量較DM2組低,差異有統計學意義(P<0.05),Glu/GABA比值與DM2組比較,差異無統計學意義(t=1.199,P>0.05)(表 1)。
RT-PCR檢測結果顯示,NC組、DM1組、MM組、DM2組大鼠視網膜組織中GAD mRNA相對表達量分別為0.577±0.069、0.472±0.051、0.409±0.027、0.311±0.024。MM組、DM1組、DM2組大鼠視網膜中GAD mRNA相對表達量均較NC組低,差異有統計學意義(F=57.668,P<0.05);MM組大鼠視網膜中GAD mRNA相對表達量較DM1組低(t=3.496, P<0.05),較DM2組高(t=8.613, P<0.05),差異有統計學意義(圖 2, 3)。
圖2
各組細胞大鼠視網膜組織中GAD mRNA相對表達量電泳圖。M:Marker;1:NC組;2~3:DM1組;4~5:MM組;6~7:DM2組
圖3
各組大鼠視網膜組織中GAD mRNA相對表達量比較。a與NC組比較,P<0.05;b與DM1組比較,P<0.05;c與DM2組比較,P<0.05
TUNEL法檢測結果顯示,NC組大鼠視網膜組織中凋亡細胞細胞核呈棕褐色,點狀分布于神經節細胞層(GCL)、內核層(INL);MM組、DM1組、DM2組大鼠視網膜中凋亡細胞顯著增多,DM2組較DM1組增多更明顯。MM組大鼠視網膜中凋亡細胞較DM2組減少,但較NC組和DM1組仍增多(圖 4)。MM組、DM1組、DM2組凋亡指數均較NC組高,差異有統計學意義(F=37.385,P<0.05);MM組凋亡指數較DM1組高(t=2.584, P<0.05),較DM2組低(t=3.531, P<0.05),差異有統計學意義(圖 5)。
圖4
各組大鼠視網膜組織光學顯微鏡像。4A.NC組,視網膜中僅見點狀陽性細胞(白箭);4B.DM1組,視網膜GCL、INL陽性細胞數量較NC組增多、密度增大;4C.MM組,視網膜GCL、INL陽性細胞數量較DM1組增多;4D.DM2組,視網膜可見大量陽性細胞。IPL:內叢狀層,OPL:外叢狀層,ONL:外核層,PL:視錐視桿細胞層,RPE:視網膜色素上皮二氨基聯苯胺×400
圖5
各組大鼠視網膜神經細胞凋亡指數比較。a與NC組比較,P<0.05;b與DM1組比較,P<0.05;c與DM2組比較,P<0.05
3 討論
長期慢性高血糖可導致DR發生發展,然而,DR等并發癥并不能完全由后期單純控制血糖來阻止。DM控制及并發癥試驗組臨床試驗、DM干預和并發癥流行病學調查試驗[9]和Holman等[10]經過長期隨訪發現,DM患者控制血糖后短期內并不能延緩DM并發癥的進展,即血管內皮細胞似乎對早期血糖環境具有記憶能力。目前, 高糖“代謝記憶”發生機制的研究是一個熱點, 但是多數研究都是圍繞高糖“記憶”損害視網膜微血管的機制展開,對視網膜神經病變的代謝記憶研究尚少。本研究重在揭示神經遞質紊亂和神經細胞凋亡在“代謝記憶”影響DR中的作用。
STZ誘導的DM鼠, 4~5周高血糖時發現星形膠質細胞激活以及數量增加[11, 12],同時也證明在膠質細胞神經膠質纖維酸性蛋白上調和反應性膠質增生[12]。從6周開始視網膜神經節細胞(RGC)數量減少[13],高血糖6周時可以鑒定RGC和血管細胞的細胞凋亡[14]。本課題組前期預實驗發現,高血糖4周時視神經細胞病變尚不明顯,8周或12周開始胰島素治療時血糖已很難控制。故本研究以6周為視網膜視神經病變研究起點。根據Bixler等[15]方法,在DM病程6周后MM組大鼠采用胰島素強化治療,結果顯示其前6周血糖與DM組大鼠血糖均處于高水平,治療后血糖下降并維持正常水平至12周。
Glu是視網膜中最主要的興奮性神經遞質,高血糖會引起視網膜缺血缺氧,引起Glu釋放增多,同時由于缺乏三磷酸腺苷,Müller細胞清除Glu能力減弱,最終導致突觸間隙中Glu堆積并激活Glu受體,Ca2+過度內流、超載,啟動系列級聯反應引起細胞離子滲透壓和電化學性質改變,產生興奮毒性,導致神經細胞凋亡。Gu等[16]認為Glu升高可誘導半胱氨酸蛋白酶非依賴的凋亡途徑,是DM視網膜神經細胞凋亡的原因之一。相關研究發現,DM大鼠視網膜中Glu含量的升高,導致血管內皮生長因子分泌增加,視網膜新生血管增多[17, 18]。由于Glu在DR發病中具有突出的作用, 本研究在DM大鼠12周后各組大鼠視網膜行HPLC檢測,MM組和DM2組大鼠視網膜組織中Glu含量相比NC組明顯升高,且視網膜神經細胞凋亡數也明顯增多。MM組在胰島素強化治療后相比DM2組Glu含量和視網膜神經細胞凋亡數降低,但仍未恢復至正常水平,說明MM組存在高糖“代謝記憶”,血糖雖然降至正常,Glu和細胞凋亡的增加仍然存在,DR持續進展。
GABA是一種重要的抑制性神經遞質,能拮抗Glu的興奮毒性,具有中樞和視網膜抑制保護作用,通常兩者之間保持平衡,Glu/GABA比值穩定。Glu被Müller細胞膜上的谷氨酸轉運體迅速轉運入胞質,既可以由谷氨酰胺合成酶轉化為無毒的谷氨酰胺,也可以經限速酶GAD催化生成GABA。視網膜缺血時,Glu釋放增多,誘導GABA釋放增加。本研究結果也顯示,MM組、DM2組大鼠視網膜組織中GABA含量相比NC組明顯升高,MM組在胰島素強化治療后相比DM2組GABA含量部分降低,但仍未恢復至正常水平。Glu/GABA比值相比Glu含量更能反應氨基酸的興奮毒性和神經功能的異常改變。本課題組前期研究證實,在DM早期大鼠視網膜組織中GABA升高,但其升高幅度不及Glu,Glu/GABA比值仍顯著升高,氨基酸毒性超過了機體的保護作用,視網膜缺血損傷繼續發生[8]。對于Glu/GABA比值,本研究發現MM組、DM2組大鼠視網膜組織中Glu/GABA的比值相比NC組均明顯升高,MM組在胰島素強化治療后相比DM2組其比值并無明顯改變,說明胰島素強化治療對前期高血糖引起的Glu/GABA比值升高無效。GAD是合成GABA的限速酶,在Müller細胞中表達豐富,GABA合成過多可抑制GAD的表達,形成負反饋。本研究結果也證實了這點,MM組、DM2組大鼠視網膜組織中GAD表達相比NC組均明顯降低,MM組在胰島素強化治療后相比DM2組GAD表達部分升高,但仍未恢復至正常水平。
前期高血糖引起的神經遞質紊亂,經后期胰島素強化治療,只能獲得部分改善,甚至對于升高的Glu/GABA比值無任何影響,視網膜神經組織仍存在Glu過興奮毒性,視網膜神經病變繼續發生發展。Glu、GABA紊亂可能是高糖“代謝記憶”致DR的機制之一。本研究提出了高糖“代謝記憶”機制的新思路,但尚未明確遞質紊亂與高糖“記憶”引起的視網膜病變存在必然的因果關系,后續將進一步研究如何降低興奮性遞質的含量,其改變是否能改善高糖“記憶”對視網膜的損害。