引用本文: 林琳, 熊永強, 呂月娥, 童林利. 皮膚色素失禁癥合并雙眼視網膜病變一例. 中華眼底病雜志, 2015, 31(5): 493-494. doi: 10.3760/cma.j.issn.1005-1015.2015.05.024 復制
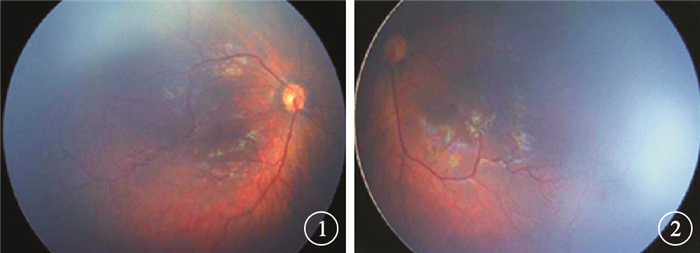
患兒女,2個月。因外院皮膚科診斷“皮膚色素失禁癥”建議行眼底檢查于2014年8月來我院眼科就診。患兒足月順產,無宮內缺氧或窒息史、吸氧史、藥物過敏史;父母非近親結婚;否認家族遺傳病史。患兒出生后軀干、四肢、腋窩及腹股溝處皮膚出現紅色斑點、夾雜小水皰,隨后部分水皰破裂、結痂,結痂處皮膚呈現褐色疣狀增生。眼科檢查:雙眼眼球運動正常,可追光;瞳孔等大等圓,直接、間接對光反射正常。眼前節檢查未見異常。指測眼壓Tn。廣角數碼視網膜成像系統檢查,雙眼玻璃體透明;視盤邊界清楚,顏色淡紅,杯盤比<0.3;黃斑中心凹反光正常;視網膜動靜脈比例1:3;顳側邊緣血管紆曲、擴張,可見出血,周邊部血管稀少,可見明顯分界線;后極部及周邊部視網膜平伏,未見滲出及視網膜脫離(圖 1, 2)。全身檢查,軀干、四肢、腋窩及腹股溝處皮膚見大片紅色斑點,部分呈現褐色疣狀增生;其他一般情況良好,牙齒未萌出,無骨骼畸形,無神經系統異常癥狀。實驗室檢查:血、尿常規正常,肝腎功能未見明顯異常。頭顱CT及腦電圖檢查結果均正常。患兒父母視力均正常;眼底檢查未見明顯異常。診斷:皮膚色素失禁癥合并雙眼視網膜病變。
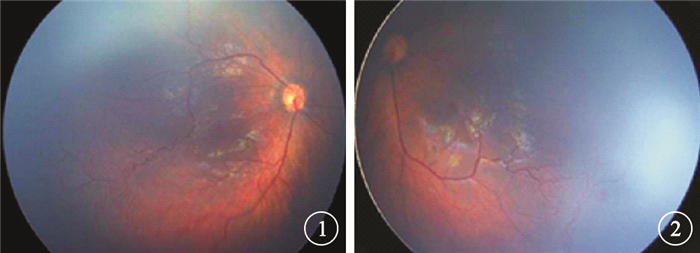 圖1
右眼彩色眼底像。右眼視網膜動靜脈比例1:3,顳側周邊血管紆曲、擴張,周邊部血管稀少,可見明顯分界線??圖 2?左眼彩色眼底像。視網膜動靜脈比例1:3,顳側周邊血管紆曲、擴張,可見出血;周邊部血管稀少,可見明顯分界線
圖1
右眼彩色眼底像。右眼視網膜動靜脈比例1:3,顳側周邊血管紆曲、擴張,周邊部血管稀少,可見明顯分界線??圖 2?左眼彩色眼底像。視網膜動靜脈比例1:3,顳側周邊血管紆曲、擴張,可見出血;周邊部血管稀少,可見明顯分界線
討論????色素失禁癥又稱Bloch-sulzberger病或Blcoh-si-ments病綜合征,是一種少見的X性連鎖顯性遺傳性疾病, 多見于女性,常有近親結婚及家族遺傳史;皮膚發生紅斑、水皰、疣狀或炎癥改變后出現色素性皮損,可伴有眼、骨和中樞神經系統缺陷[1]。Carney[2]報道的445例患者中,35.2%的患者有眼部改變。其中,單眼或雙眼失明者34例,占7.5%;嚴重眼部異常者86例,占18.9%。認為眼部病變是本病最嚴重的并發癥。眼部病變常見表現為視網膜血管異常、視網膜脫離、視網膜色素上皮改變等視網膜病變;其次為斜視、白內障、視神經萎縮等,受累眼常表現為小眼球[3]。患兒出生后眼底即可出現毛細血管擴張、出血,黃斑區缺血性梗阻,動靜脈吻合,周邊視網膜低灌注區,繼發性新生血管、視網膜增生牽拉視網膜脫離,視網膜前纖維組織形成,晶狀體后團塊形成等[4]。
本例患兒皮膚出現紅斑及大皰、丘疹和疣狀損害及色素沉著等改變,符合皮膚色素失禁癥臨床表現[5]。眼底視網膜血管紆曲、擴張,動靜脈比例1:3,周邊部血管極少,可見明顯分界線,視網膜病變診斷確立。本病應與早產兒視網膜病變、家族性滲出性玻璃體視網膜病變相鑒別。本例患兒其視網膜病變特征與早產兒視網膜病變早期改變相似,但患兒為足月兒,體重正常,無吸氧史,故可排除。患兒父母眼底檢查未見明顯異常,結合患兒皮膚病變特點,亦可排除家族性滲出性玻璃體視網膜病變。
眼部病變可在任一階段停止,遺留視網膜無血管區、血管紆曲、玻璃體積血等,并長期穩定,大部分眼底無血管灌注區并不需要治療[6]。對進展性視網膜新生血管,可采用激光光凝或冷凍治療。近年來玻璃體腔注射抗血管內皮生長因子藥物治療色素失禁癥眼部病變亦取得較好療效[7]。本例患兒視網膜病變為早期病變, 因此未進行干預。但針對此類患兒應定期隨訪,若出現視網膜脫離可早期干預;而出現視網膜全脫離則失去治療時機[8, 9]。
患兒女,2個月。因外院皮膚科診斷“皮膚色素失禁癥”建議行眼底檢查于2014年8月來我院眼科就診。患兒足月順產,無宮內缺氧或窒息史、吸氧史、藥物過敏史;父母非近親結婚;否認家族遺傳病史。患兒出生后軀干、四肢、腋窩及腹股溝處皮膚出現紅色斑點、夾雜小水皰,隨后部分水皰破裂、結痂,結痂處皮膚呈現褐色疣狀增生。眼科檢查:雙眼眼球運動正常,可追光;瞳孔等大等圓,直接、間接對光反射正常。眼前節檢查未見異常。指測眼壓Tn。廣角數碼視網膜成像系統檢查,雙眼玻璃體透明;視盤邊界清楚,顏色淡紅,杯盤比<0.3;黃斑中心凹反光正常;視網膜動靜脈比例1:3;顳側邊緣血管紆曲、擴張,可見出血,周邊部血管稀少,可見明顯分界線;后極部及周邊部視網膜平伏,未見滲出及視網膜脫離(圖 1, 2)。全身檢查,軀干、四肢、腋窩及腹股溝處皮膚見大片紅色斑點,部分呈現褐色疣狀增生;其他一般情況良好,牙齒未萌出,無骨骼畸形,無神經系統異常癥狀。實驗室檢查:血、尿常規正常,肝腎功能未見明顯異常。頭顱CT及腦電圖檢查結果均正常。患兒父母視力均正常;眼底檢查未見明顯異常。診斷:皮膚色素失禁癥合并雙眼視網膜病變。
圖1
右眼彩色眼底像。右眼視網膜動靜脈比例1:3,顳側周邊血管紆曲、擴張,周邊部血管稀少,可見明顯分界線??圖 2?左眼彩色眼底像。視網膜動靜脈比例1:3,顳側周邊血管紆曲、擴張,可見出血;周邊部血管稀少,可見明顯分界線
討論????色素失禁癥又稱Bloch-sulzberger病或Blcoh-si-ments病綜合征,是一種少見的X性連鎖顯性遺傳性疾病, 多見于女性,常有近親結婚及家族遺傳史;皮膚發生紅斑、水皰、疣狀或炎癥改變后出現色素性皮損,可伴有眼、骨和中樞神經系統缺陷[1]。Carney[2]報道的445例患者中,35.2%的患者有眼部改變。其中,單眼或雙眼失明者34例,占7.5%;嚴重眼部異常者86例,占18.9%。認為眼部病變是本病最嚴重的并發癥。眼部病變常見表現為視網膜血管異常、視網膜脫離、視網膜色素上皮改變等視網膜病變;其次為斜視、白內障、視神經萎縮等,受累眼常表現為小眼球[3]。患兒出生后眼底即可出現毛細血管擴張、出血,黃斑區缺血性梗阻,動靜脈吻合,周邊視網膜低灌注區,繼發性新生血管、視網膜增生牽拉視網膜脫離,視網膜前纖維組織形成,晶狀體后團塊形成等[4]。
本例患兒皮膚出現紅斑及大皰、丘疹和疣狀損害及色素沉著等改變,符合皮膚色素失禁癥臨床表現[5]。眼底視網膜血管紆曲、擴張,動靜脈比例1:3,周邊部血管極少,可見明顯分界線,視網膜病變診斷確立。本病應與早產兒視網膜病變、家族性滲出性玻璃體視網膜病變相鑒別。本例患兒其視網膜病變特征與早產兒視網膜病變早期改變相似,但患兒為足月兒,體重正常,無吸氧史,故可排除。患兒父母眼底檢查未見明顯異常,結合患兒皮膚病變特點,亦可排除家族性滲出性玻璃體視網膜病變。
眼部病變可在任一階段停止,遺留視網膜無血管區、血管紆曲、玻璃體積血等,并長期穩定,大部分眼底無血管灌注區并不需要治療[6]。對進展性視網膜新生血管,可采用激光光凝或冷凍治療。近年來玻璃體腔注射抗血管內皮生長因子藥物治療色素失禁癥眼部病變亦取得較好療效[7]。本例患兒視網膜病變為早期病變, 因此未進行干預。但針對此類患兒應定期隨訪,若出現視網膜脫離可早期干預;而出現視網膜全脫離則失去治療時機[8, 9]。