引用本文: 曹葭, 孫尉, 邵珺, 謝田華, 殷麗, 吳瑩, 王如興, 姚勇. 糖尿病大鼠視網膜動脈平滑肌細胞大電導鈣離子激活鉀離子通道開放概率及其亞基蛋白表達. 中華眼底病雜志, 2015, 31(2): 162-164. doi: 10.3760/cma.j.issn.1005-1015.2015.02.013 復制
糖尿病(DM)發病時視網膜血流動力學異常早于DM視網膜病變(DR)的發生,并且與其病變進展密切相關[1-3]。DR前期視網膜中央動脈及其分支的異常收縮,可導致視網膜血流灌注減少,引起視網膜組織缺血缺氧。而視網膜動脈離子通道的異常是導致血管異常收縮、血流動力學紊亂的主要原因之一,其中視網膜動脈平滑肌細胞(RVSMCs)大電導鈣離子激活鉀離子(BK)通道的改變尤為重要[4, 5]。為了探尋RVSMCs的BK通道在DR發生發展中的作用,我們觀察了DM大鼠RVSMCs的BK通道總開放概率(NP0)及α、β1亞基相對蛋白表達。現將結果報道如下。
1 材料和方法
8~12周齡健康雄性Spraque-Dawley大鼠50只,體重約(200±30)g,江蘇省血吸蟲病防治研究所動物中心提供。將大鼠隨機分為正常對照組、DM模型組,分別為10、40只。DM模型組大鼠以60 mg/kg的劑量腹腔注射鏈脲佐菌素建模,以最終血糖濃度連續8周大于16.7 mmol/L為建模成功[6]。正常對照組大鼠腹腔注射等劑量0.9%生理鹽水作為對照。常規麻醉處死大鼠,摘取保存眼球,四步酶消化法分離RVSMCs[7]。最后將消化的組織用保存液漂洗2~3次后,放入保存液中4℃靜置數小時,使用前輕吹打,得到大量RVSMCs。采用倒置顯微鏡觀察RVSMCs形態,輪廓清晰、邊緣不毛糙細胞為存活細胞。
采用Axopatch 200B膜片鉗放大器和pCLAMP 10.2軟件記錄BK單通道電流。以膜內向外型模式記錄BK單通道電流。輸出信號經8極Bessel濾波器濾波,采樣頻率20 kHz,濾波頻率5 kHz,電極電阻5~10 M。電極內液: KCl 140.0 mmol/L、4-羥乙基哌嗪乙磺酸(HEPES) 10.0 mmol/L、乙二醇雙四乙酸(EGTA)1.0 mmol/L、MgCl2 1.0μmol/L、CaCl2 1.0μmol/L,pH7.4;電極外液: KCl 140.0 mmol/L、HEPES 10.0 mmol/L、EGTA 1.0 mmol/L、MgCl2 1.0μmol/L,pH7.35。在刺激電位為0、20、40、60、80、100、120 mV條件下,分別記錄正常對照組及DM模型組大鼠RVSMCs的BK通道NP0,比較其變化。采用公式NP0=∑(Onn)/T計算BK通道NP0[8]。其中,N為通道的開放數量,P0代表開放概率,T代表總記錄時間,On代表每個開放水平所需時間。
采用蛋白免疫印跡法(Western blot)檢測大鼠RVSMCs中α、β1亞基的相對蛋白表達。分離大鼠視網膜動脈,血管組織經液氮速凍后于-80℃儲存備用。取出凍存大鼠視網膜動脈組織加入RIPA裂解液,在冰上充分研磨至無組織塊,以13 000×g,4℃條件下離心15 min,離心后收集上清液,采用二喹啉甲酸法對提取的蛋白質進行濃度測定。等量蛋白分別行7%(α亞基,相對分子質量約125×103)和12%(β1亞基,相對分子質量約28×103)丙烯酰胺凝膠電泳,5%脫脂牛奶室溫封閉2 h,抗BK通道α亞基和β1亞基抗體4℃孵育過夜,洗膜,辣根過氧化物酶標記二抗室溫孵育2 h洗膜后再用電化學發光試劑盒發光壓片,掃描膠片后吸光度[A, 舊稱光密度(OD)]分析法定量。磷酸甘油醛脫氫酶(GAPDH)作為內參。
采用SPSS 18.0統計軟件行統計學分析。計量資料用均數±標準差(
2 結果
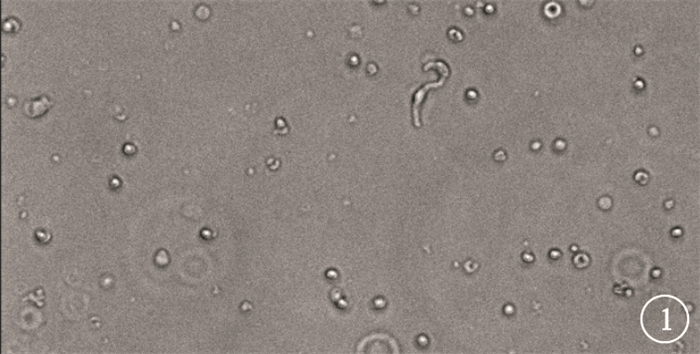
倒置顯微鏡觀察發現,RVSMCs可呈蚯蚓狀、短桿狀、花生狀,形態各異(圖 1)。
 圖1
正常RVSMCs倒置顯微鏡像。RVSMCs可呈蚯蚓狀、短桿狀、花生狀×200
圖1
正常RVSMCs倒置顯微鏡像。RVSMCs可呈蚯蚓狀、短桿狀、花生狀×200
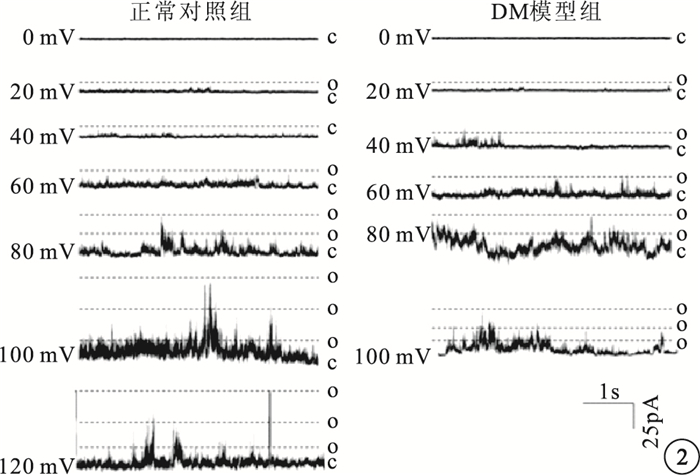
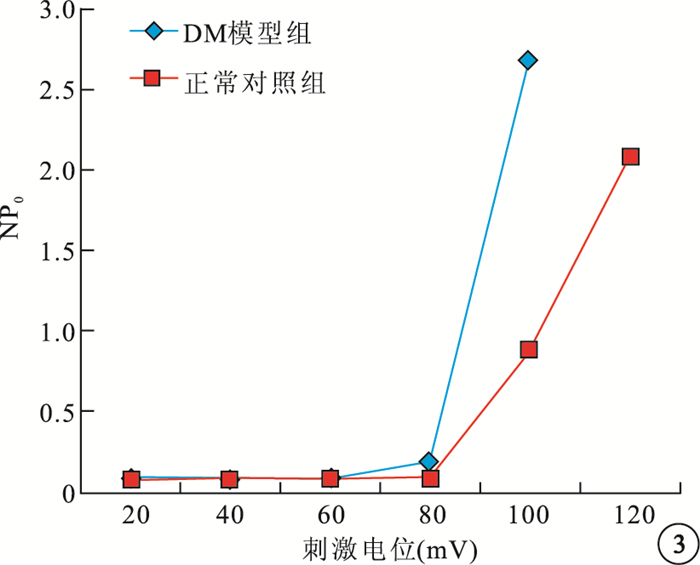
0~120 mV不同刺激電壓下,DM模型組大鼠RVSMCs的BK通道NP0明顯增高,差異有統計學意義(t=4.260,P<0.05)(圖 2,3)。
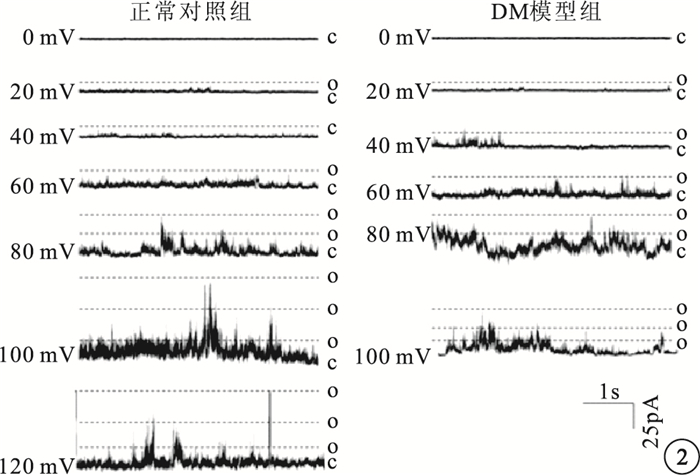 圖2
兩組大鼠RVSMCs的BK通道電流。O:通道開放;C:通道關閉
圖2
兩組大鼠RVSMCs的BK通道電流。O:通道開放;C:通道關閉
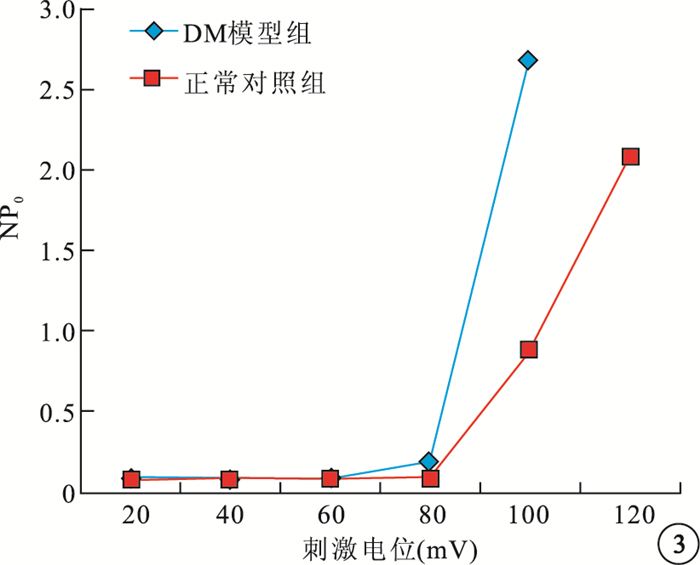 圖3
兩組大鼠RVSMCs的BK通道電壓NP0折線圖
圖3
兩組大鼠RVSMCs的BK通道電壓NP0折線圖
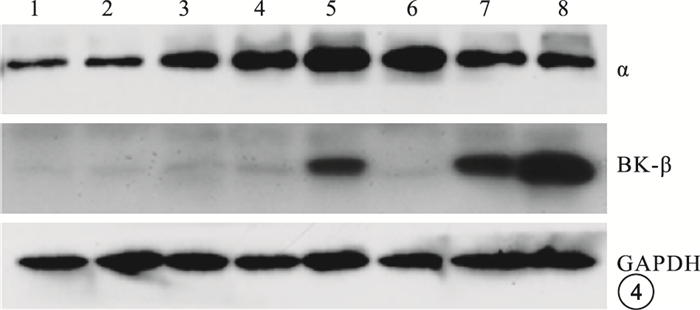
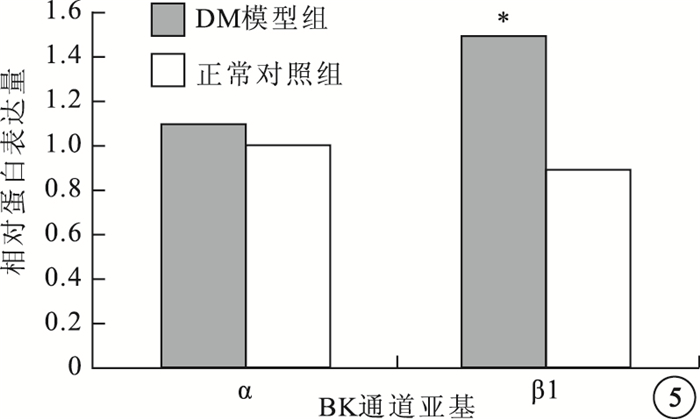
與正常對照組比較,DM模型組大鼠RVSMCs的BK通道α亞基蛋白表達無明顯變化,差異無統計學意義(t=10.126,P>0.05);β1亞基蛋白表達明顯升高,差異有統計學意義(t=5.146,P<0.05)(圖 4,5)。
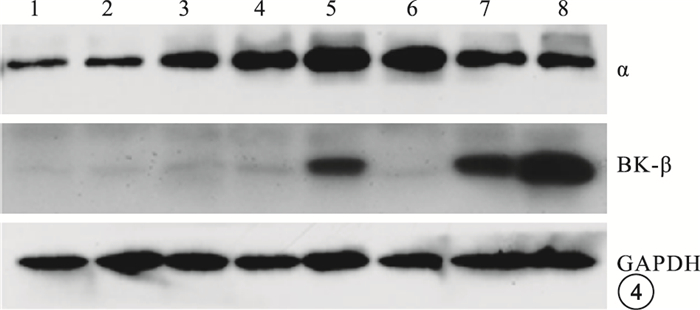 圖4
兩組大鼠RVSMCs的BK通道α、β1亞基蛋白電泳圖。1~4為正常對照組,5~8為DM模型組
圖4
兩組大鼠RVSMCs的BK通道α、β1亞基蛋白電泳圖。1~4為正常對照組,5~8為DM模型組
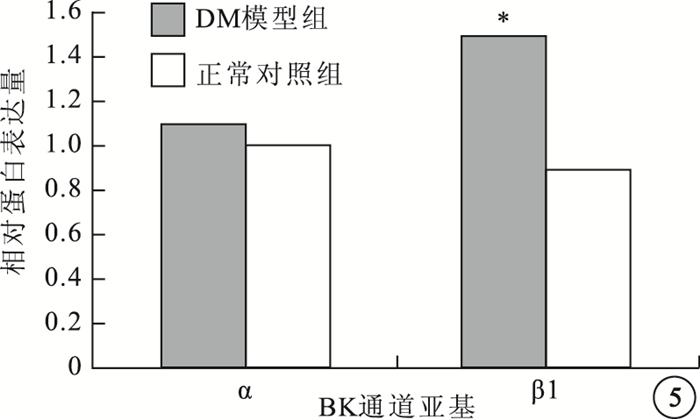 圖5
兩組大鼠RVSMCs的BK通道α、β1亞基蛋白表達比較。*與正常對照組比較,P<0.05
圖5
兩組大鼠RVSMCs的BK通道α、β1亞基蛋白表達比較。*與正常對照組比較,P<0.05
3 討論
BK通道廣泛存在于興奮及非興奮細胞中,參與包括動作電位復極化、神經遞質釋放、激素分泌以及血管張力調節等生理活動[9]。BK通道是由α亞單位及β亞單位組成的四聚體結構[10]。α亞單位由單基因KCNMAl編碼,廣泛存在于哺乳動物組織中,并且在種屬之間表現出高度同源性,其是形成BK離子通道的部位;β亞單位由KCNMB l~4編碼,有4種亞型,在血管平滑肌細胞上主要為β1亞單位;β亞單位通過與α亞單位相互作用,可以改變BK通道的電壓依賴性及鈣敏感性。正常情況下,BK通道激活后鉀離子外流,產生超極化,抑制電壓依賴性鈣離子通道開放,進而鈣離子內流減少,引起血管平滑肌舒張,故BK通道功能主要為參與血管的擴張[11]。有研究發現,DM及DM前期病變時,由BK通道介導的血管擴張功能受損,但其確切分子機制尚不十分清楚,DM時BK通道α亞基及β1亞基蛋白表達是上調亦或是下降尚需進一步研究證實[12-14]。
本研究結果顯示,與正常對照組比較,DM模型組大鼠RVSMCs的NP0明顯升高,與McGahon等[15]報道結果相反。我們分析是以下兩方面的原因引起:(1)國外研究多以分離并消化后仍位于視網膜動脈小片段上的平滑肌細胞為研究對象,本研究則采用急性酶消化法分離的單個RVSMCs為研究對象,可能造成其電生理特點不同,進而引起本次實驗結果與國外報道結果的不一致。(2)我們在建模后8周便處死了大鼠進行相關實驗,研究的是DM早期病變。有研究表明,DR一般在建模24周后才會出現,而本研究并未發現大鼠眼底血管出現明顯DR的表現。因此,我們推測在DR發生前期,BK通道NP0增加,可能是作為一種保護性代償機制,以抵抗DM引起的微血管病變。
本研究結果顯示,與正常對照組比較,DM模型組大鼠RVSMCs的BK通道α亞基蛋白表達無明顯變化,而β1亞基蛋白表達明顯升高。與McGahon等[15]報道結果一致。該Western blot檢測結果與BK通道NP0增加相一致。說明在DR發生前期,RVSMCs的BK通道出現代償性增加的機制,可能通過BK通道β1亞基蛋白表達上調,導致了NP0升高,代償性抵抗了DR的早期發生。
本研究結果表明,DM早期RVSMCs的BK通道β1亞基蛋白表達升高,BK通道NP0增加,可能是作為一種保護性代償機制抵抗DR的早期發生。在國內外研究的基礎上,本研究拓展了DR發生機制,提出了更具前瞻性的假設,為尋找可能的藥物作用靶點預防及治療早期DR提供了新思路。
糖尿病(DM)發病時視網膜血流動力學異常早于DM視網膜病變(DR)的發生,并且與其病變進展密切相關[1-3]。DR前期視網膜中央動脈及其分支的異常收縮,可導致視網膜血流灌注減少,引起視網膜組織缺血缺氧。而視網膜動脈離子通道的異常是導致血管異常收縮、血流動力學紊亂的主要原因之一,其中視網膜動脈平滑肌細胞(RVSMCs)大電導鈣離子激活鉀離子(BK)通道的改變尤為重要[4, 5]。為了探尋RVSMCs的BK通道在DR發生發展中的作用,我們觀察了DM大鼠RVSMCs的BK通道總開放概率(NP0)及α、β1亞基相對蛋白表達。現將結果報道如下。
1 材料和方法
8~12周齡健康雄性Spraque-Dawley大鼠50只,體重約(200±30)g,江蘇省血吸蟲病防治研究所動物中心提供。將大鼠隨機分為正常對照組、DM模型組,分別為10、40只。DM模型組大鼠以60 mg/kg的劑量腹腔注射鏈脲佐菌素建模,以最終血糖濃度連續8周大于16.7 mmol/L為建模成功[6]。正常對照組大鼠腹腔注射等劑量0.9%生理鹽水作為對照。常規麻醉處死大鼠,摘取保存眼球,四步酶消化法分離RVSMCs[7]。最后將消化的組織用保存液漂洗2~3次后,放入保存液中4℃靜置數小時,使用前輕吹打,得到大量RVSMCs。采用倒置顯微鏡觀察RVSMCs形態,輪廓清晰、邊緣不毛糙細胞為存活細胞。
采用Axopatch 200B膜片鉗放大器和pCLAMP 10.2軟件記錄BK單通道電流。以膜內向外型模式記錄BK單通道電流。輸出信號經8極Bessel濾波器濾波,采樣頻率20 kHz,濾波頻率5 kHz,電極電阻5~10 M。電極內液: KCl 140.0 mmol/L、4-羥乙基哌嗪乙磺酸(HEPES) 10.0 mmol/L、乙二醇雙四乙酸(EGTA)1.0 mmol/L、MgCl2 1.0μmol/L、CaCl2 1.0μmol/L,pH7.4;電極外液: KCl 140.0 mmol/L、HEPES 10.0 mmol/L、EGTA 1.0 mmol/L、MgCl2 1.0μmol/L,pH7.35。在刺激電位為0、20、40、60、80、100、120 mV條件下,分別記錄正常對照組及DM模型組大鼠RVSMCs的BK通道NP0,比較其變化。采用公式NP0=∑(Onn)/T計算BK通道NP0[8]。其中,N為通道的開放數量,P0代表開放概率,T代表總記錄時間,On代表每個開放水平所需時間。
采用蛋白免疫印跡法(Western blot)檢測大鼠RVSMCs中α、β1亞基的相對蛋白表達。分離大鼠視網膜動脈,血管組織經液氮速凍后于-80℃儲存備用。取出凍存大鼠視網膜動脈組織加入RIPA裂解液,在冰上充分研磨至無組織塊,以13 000×g,4℃條件下離心15 min,離心后收集上清液,采用二喹啉甲酸法對提取的蛋白質進行濃度測定。等量蛋白分別行7%(α亞基,相對分子質量約125×103)和12%(β1亞基,相對分子質量約28×103)丙烯酰胺凝膠電泳,5%脫脂牛奶室溫封閉2 h,抗BK通道α亞基和β1亞基抗體4℃孵育過夜,洗膜,辣根過氧化物酶標記二抗室溫孵育2 h洗膜后再用電化學發光試劑盒發光壓片,掃描膠片后吸光度[A, 舊稱光密度(OD)]分析法定量。磷酸甘油醛脫氫酶(GAPDH)作為內參。
采用SPSS 18.0統計軟件行統計學分析。計量資料用均數±標準差(
2 結果
倒置顯微鏡觀察發現,RVSMCs可呈蚯蚓狀、短桿狀、花生狀,形態各異(圖 1)。
圖1
正常RVSMCs倒置顯微鏡像。RVSMCs可呈蚯蚓狀、短桿狀、花生狀×200
0~120 mV不同刺激電壓下,DM模型組大鼠RVSMCs的BK通道NP0明顯增高,差異有統計學意義(t=4.260,P<0.05)(圖 2,3)。
圖2
兩組大鼠RVSMCs的BK通道電流。O:通道開放;C:通道關閉
圖3
兩組大鼠RVSMCs的BK通道電壓NP0折線圖
與正常對照組比較,DM模型組大鼠RVSMCs的BK通道α亞基蛋白表達無明顯變化,差異無統計學意義(t=10.126,P>0.05);β1亞基蛋白表達明顯升高,差異有統計學意義(t=5.146,P<0.05)(圖 4,5)。
圖4
兩組大鼠RVSMCs的BK通道α、β1亞基蛋白電泳圖。1~4為正常對照組,5~8為DM模型組
圖5
兩組大鼠RVSMCs的BK通道α、β1亞基蛋白表達比較。*與正常對照組比較,P<0.05
3 討論
BK通道廣泛存在于興奮及非興奮細胞中,參與包括動作電位復極化、神經遞質釋放、激素分泌以及血管張力調節等生理活動[9]。BK通道是由α亞單位及β亞單位組成的四聚體結構[10]。α亞單位由單基因KCNMAl編碼,廣泛存在于哺乳動物組織中,并且在種屬之間表現出高度同源性,其是形成BK離子通道的部位;β亞單位由KCNMB l~4編碼,有4種亞型,在血管平滑肌細胞上主要為β1亞單位;β亞單位通過與α亞單位相互作用,可以改變BK通道的電壓依賴性及鈣敏感性。正常情況下,BK通道激活后鉀離子外流,產生超極化,抑制電壓依賴性鈣離子通道開放,進而鈣離子內流減少,引起血管平滑肌舒張,故BK通道功能主要為參與血管的擴張[11]。有研究發現,DM及DM前期病變時,由BK通道介導的血管擴張功能受損,但其確切分子機制尚不十分清楚,DM時BK通道α亞基及β1亞基蛋白表達是上調亦或是下降尚需進一步研究證實[12-14]。
本研究結果顯示,與正常對照組比較,DM模型組大鼠RVSMCs的NP0明顯升高,與McGahon等[15]報道結果相反。我們分析是以下兩方面的原因引起:(1)國外研究多以分離并消化后仍位于視網膜動脈小片段上的平滑肌細胞為研究對象,本研究則采用急性酶消化法分離的單個RVSMCs為研究對象,可能造成其電生理特點不同,進而引起本次實驗結果與國外報道結果的不一致。(2)我們在建模后8周便處死了大鼠進行相關實驗,研究的是DM早期病變。有研究表明,DR一般在建模24周后才會出現,而本研究并未發現大鼠眼底血管出現明顯DR的表現。因此,我們推測在DR發生前期,BK通道NP0增加,可能是作為一種保護性代償機制,以抵抗DM引起的微血管病變。
本研究結果顯示,與正常對照組比較,DM模型組大鼠RVSMCs的BK通道α亞基蛋白表達無明顯變化,而β1亞基蛋白表達明顯升高。與McGahon等[15]報道結果一致。該Western blot檢測結果與BK通道NP0增加相一致。說明在DR發生前期,RVSMCs的BK通道出現代償性增加的機制,可能通過BK通道β1亞基蛋白表達上調,導致了NP0升高,代償性抵抗了DR的早期發生。
本研究結果表明,DM早期RVSMCs的BK通道β1亞基蛋白表達升高,BK通道NP0增加,可能是作為一種保護性代償機制抵抗DR的早期發生。在國內外研究的基礎上,本研究拓展了DR發生機制,提出了更具前瞻性的假設,為尋找可能的藥物作用靶點預防及治療早期DR提供了新思路。