引用本文: 魯理, 鄭志, 李靜文, 肖文瑋, 李春霞. 高糖誘導的牛視網膜血管內皮細胞中共濟失調毛細血管擴張突變基因激酶活性變化及其對細胞氧化應激狀態的影響. 中華眼底病雜志, 2014, 30(6): 604-607. doi: 10.3760/cma.j.issn.1005-1015.2014.06.016 復制
氧化應激與視網膜組織的炎癥反應、神經血管損害、細胞凋亡、線粒體功能紊亂等聯系密切,在糖尿病視網膜病變(DR)的病理過程中起重要作用[1-3]。共濟失調毛細血管擴張突變基因(ATM)激酶作為一種DNA損傷修復過程中的經典蛋白分子,在氧化應激中被激活并起到調控氧化應激的作用[4]。ATM缺失導致缺氧誘導因子1(HIF-1)大量增加從而加劇細胞氧化應激[5]。但目前有關ATM激酶在DR發病機制中的確切作用還不明確。為此,我們觀察高糖培養狀態下體外培養的牛視網膜微血管內皮細胞(BRECs)中ATM激酶的活性和表達變化及其對氧化應激的調節作用,初步探究ATM激酶在DR中的作用機制,為防治DR提供新的思路。現將結果報道如下。
1 材料和方法
1.1 BRECs培養及實驗分組
參照文獻[5, 6]的選擇性培養方法培養BRECs。取新鮮獲得的牛眼球,取出并剪碎視網膜微血管,加入含0.05%的膠原酶和0.025%的牛血清蛋白消化液,37℃消化45 min。應用網孔直徑88 μm的不銹鋼篩網過濾,D-hank液沖洗濾網上的組織,在離心后的小丸狀物中加入含有10%滅活胎牛血清、100 μg/ml肝素、15 μg/ml內皮細胞生長添加物、10 mmol/L羥乙基哌嗪乙磺酸的Dulbecco改良Eagle培養基,接種于包被有明膠的培養皿培養。傳代后,取第1代細胞,通過vonWillebrand因子抗體,免疫細胞化學方法對內皮細胞進行鑒定。取4~8代的細胞,傳代培養24 h后分為正常糖組、高糖組、干預組進行實驗。分別采用含5 mmol/L葡萄糖、30 mmol/L葡萄糖、30 mmol/L葡萄糖和10 μmol/L ATM激酶特異性抑制劑KU-55933的細胞培養液繼續培養48 h進行相關實驗。
1.2 實驗方法及檢測指標
細胞培養后48 h,采用蛋白免疫印跡法(Western blot)檢測細胞中ATM、磷酸化ATM(P-ATM)、細胞分裂素活化蛋白激酶(P38)、磷酸化P38(P-P38)、細胞外信號調節激酶(ERK)1/2、磷酸化ERK1/2(P-ERK1/2)的表達。應用二喹啉甲酸蛋白定量法測定裂解液提取各組細胞總蛋白。按照蛋白定量結果,以4:1的比例混合蛋白樣品及5倍上樣緩沖液,100℃加熱3~5 min,按每孔20 μg蛋白量上樣,進行電泳、轉膜、封閉、孵育一抗與二抗,包括單克隆兔抗β微管蛋白抗體、多克隆兔抗P38抗體、多克隆兔抗P-P38抗體、多克隆兔抗ERK1/2抗體、多克隆兔抗P-ERK1/2抗體(美國CST公司),辣根過氧化物酶羊抗兔IgG二抗(美國Bioworld公司),單克隆小鼠抗ATM抗體、單克隆小鼠抗P-ATM抗體(英國Abcam公司)及ATM激酶抑制劑KU55933(美國R & D公司),最后顯影定影。采用Image J圖像處理軟件進行灰度分析,以磷酸化蛋白與其總蛋白的比值表示相應蛋白的活性水平。
細胞培養后48 h,采用酶聯免疫吸附試驗(ELISA)檢測細胞上清液中血管內皮生長因子(VEGF)的含量。收集各組細胞上清液,每孔100 μl,設復孔,按照人VEGF ELISA試劑盒(上海依科賽公司)說明書進行操作。用酶聯免疫檢測儀在波長450 nm下測量吸光度[A,舊稱光密度(OD)]值,獲取細胞上清液中VEGF的含量。
細胞培養后48 h,采用細胞內ROS水平檢測試劑盒檢測細胞內ROS的水平。細胞培養后48 h,采用碘化丙啶(PI)和赫斯特染料(Hoechst)雙染色法檢測細胞凋亡情況。收集對數生長期細胞,以細胞密度2×105個細胞/孔接種于24孔培養板中。分別稱取10 mg PI或Hoechst溶于10 ml磷酸鹽緩沖液(PBS)中,終濃度為1 mg/ml。分裝后PI 4℃避光儲存,Hoechst 20℃避光儲存。配制工作液,PI:Hoechst:PBS體積比為1:1:100。吸去上清液,每孔加入工作液500 μl,置于培養箱避光培養15 min。采用熒光顯微鏡觀察細胞,以PI著紅色細胞為壞死及凋亡晚期細胞,Hoechst著藍色細胞為全部細胞,以每100個細胞中著紅色細胞數/著藍色細胞數之比為細胞凋亡率[5]。
細胞培養后48 h,終止培養,原位裝載探針,采用異硫氰酸熒光素標記的葡聚糖檢測內皮細胞屏障的通透性。按照1:1000的比例,用無血清培養液稀釋2′, 7′-二氯熒光二乙酸鹽(DCFH-DA),使終濃度為10 μmol/L。去除細胞培養液,用無血清培養液洗滌1次,加入適當體積無血清培養基稀釋好的DCFH-DA,加入的體積以能充分蓋住細胞為準。37℃細胞培養箱內孵育25~30 min。PBS洗滌細胞3次,充分去除未進入細胞內的DCFH-DA。應用熒光顯微鏡觀察并拍照。采用Image J圖像處理軟件進行定量分析。將BRECs接種于Transwell微孔膜上(美國Costar公司),微孔膜直徑24 mm,孔徑0.4 μm,參照文獻[7]的方法計算處理各組細胞旁通透率。
1.3 統計學方法
采用SPSS 17.0統計學軟件進行統計學分析。計量資料數據以均數±標準差(
2 結果
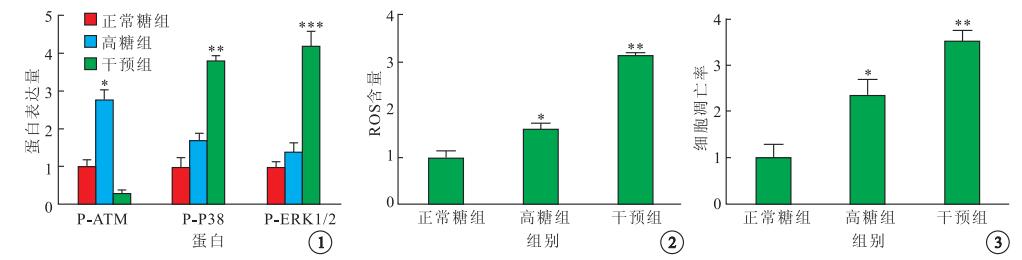
Western blot檢測結果顯示,正常糖組、高糖組及干預組P-ATM、P-P38、P-ERK1/2蛋白表達比較,差異均有統計學意義(F=436.00、14 010.00、985.10,P<0.05)。與正常糖組比較,高糖組P-ATM、P-P38、P-ERK1/2蛋白表達增高,差異均有統計學意義(t=-9.74、-4.70、-20.33,P<0.05)。與高糖組比較,干預組P-ATM蛋白表達降低,差異有統計學意義(t=5.44,P<0.01);p-p38、p-ERK1/2蛋白表達明顯增加,差異均有統計學意義(t=-18.37、-12.97,P<0.01)(圖 1)。
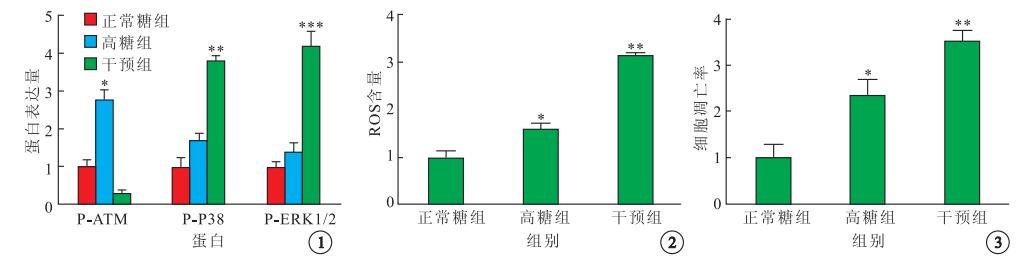 圖1
各組P-ATM、P-P38及P-ERK1/2蛋白表達比較。*高糖組與正常糖組比較,P<0.05;**干預組與高糖組比較,P<0.05;***干預組與高糖組比較,P<0.05??圖 2??各組BRECs中ROS含量比較。*高糖組與正常糖組比較,P<0.05;**干預組與高糖組比較,P<0.05??圖 3??各組BRECs細胞凋亡率比較。*高糖組與正常糖組比較,P<0.05;**干預組與高糖組比較,P<0.05
圖1
各組P-ATM、P-P38及P-ERK1/2蛋白表達比較。*高糖組與正常糖組比較,P<0.05;**干預組與高糖組比較,P<0.05;***干預組與高糖組比較,P<0.05??圖 2??各組BRECs中ROS含量比較。*高糖組與正常糖組比較,P<0.05;**干預組與高糖組比較,P<0.05??圖 3??各組BRECs細胞凋亡率比較。*高糖組與正常糖組比較,P<0.05;**干預組與高糖組比較,P<0.05
ELISA檢測結果顯示,正常糖組、高糖組、干預組細胞上清液中VEGF含量分別為(138.0±4.6)、(210.0±6.5)、(305.0±7.1) pg/ml,3組間差異有統計學意義(F=278.00,P<0.05)。與正常糖組比較,高糖組細胞上清液中VEGF含量有所增加,差異有統計學意義(t=-13.90,P<0.05)。與高糖組比較,干預組細胞上清液中VEGF含量明顯增加,差異也有統計學意義(t=-32.79,P<0.05)。
ROS含量檢測結果顯示,與正常糖組比較,高糖組ROS含量增高約1.5倍,差異有統計學意義(t=2.89,P<0.05)。與高糖組比較,干預組ROS含量增高約3.0倍,差異也有統計學意義(t=10.91,P<0.05)(圖 2)。
PI和Hoechst雙染色法檢測結果顯示,高糖組BRECs細胞凋亡率較正常糖組明顯增加,而又明顯低于干預組,差異均有統計學意義(t=4.31、11.14,P<0.05)。3組間BRECs細胞凋亡率比較,差異有統計學意義(F=55.51,P<0.05)(圖 3)。
正常糖組、高糖組、干預組細胞旁通透率分別為(1.12±0.13)×10-6、(1.45±0.17)×10-6、(1.66±0.12)×10-6 cm/s。與正常糖組比較,高糖組、干預組細胞旁通透率均升高,以干預組升高更明顯。3組間細胞旁通透率比較,差異有統計學意義(F=2 223.00,P<0.05)。
3 討論
本研究選取體外培養的BRECs,分別以正常濃度葡萄糖、高濃度葡萄糖以及高濃度葡萄糖聯合ATM激酶抑制劑處理細胞,觀察在不同葡萄糖濃度環境中ATM激酶的激活和表達情況、細胞內氧化應激水平、細胞凋亡水平以及細胞旁通透率的變化。結果表明,高糖組BRECs的氧化應激水平明顯比正常糖組提高,ROS激增,P-ATM激酶表達增加。說明ATM激酶在高糖環境中因為氧化應激而被ROS激活,與國外研究結果相似[4]。而在采用高糖加KU55933的干預組中,我們發現細胞的氧化應激水平與高糖組相比有進一步的提高。表明ATM激酶在氧化應激中可能起到保護作用。我們還發現,高糖組中p-p38、p-ERK1/2表達輕度增高,而干預組中p-p38、p-ERK1/2表達則大大增加。P38和ERK通路是重要的凋亡通路,同時其與細胞內氧化應激狀態聯系密切。Lu等[8]研究發現,由水銀所導致的神經元損傷中氧化應激介導的p38、ERK1/2激活是主要的細胞凋亡途徑。Tian等[9]在研究牛蒡根的神經保護作用中也發現,通過降低ROS可以抑制p38、ERK1/2的激活及其所誘導的細胞凋亡。國外亦有相關文獻報道,在ATM基因敲除鼠的神經干細胞和星形膠質細胞中P38和ERK通路的激活,通過上調CDK抑制物和凋亡相關基因的表達,抑制了細胞的增生,誘導細胞生長停滯和凋亡[10, 11]。本研究結果同樣證實了ATM激酶的抑制導致P38和ERK通路的激活,從而增加了細胞的凋亡。據此我們推測,在BRECs中,ATM激酶缺失后,細胞內氧化應激失控,ROS爆發式產生,進而激活P38和ERK通路造成BRECs凋亡。說明ATM激酶在氧化應激中至關重要,其活性關系到視網膜血管內皮細胞凋亡與否。在DR病理變化中如能保持和提高ATM激酶的活性,控制細胞氧化應激,可能對控制DR發展起到積極作用。
高糖誘導的VEGF增高和由于高水平VEGF所導致的血視網膜屏障破壞、血管滲漏以及后期的新生血管形成都與DR病理過程密切相關。我們在探索ATM與VEGF的關系時發現,高糖組VEGF表達高于正常糖組,而干預組VEGF水平遠遠高于高糖組。我們分析認為,高糖環境下細胞處于氧化應激的狀態并產生大量ROS,其原因則是細胞能量代謝系統功能紊亂,線粒體電子傳遞鏈破壞,此時細胞無法正常利用氧進行有氧呼吸,處于一種缺氧的狀態[12]。ATM激酶作為一種對于缺氧和氧化應激的應答,起到保護性作用。當ATM激酶被抑制或敲除,細胞缺氧,氧化應激水平將進一步提高,導致VEGF上調。Ousset等[5]在敲除ATM基因的海拉細胞中發現,通過HIF-1高表達,可誘導作為其下游的VEGF表達增高。該結果與我們的推測一致。我們還發現各組細胞旁通透性測定結果與VEGF表達趨勢一致。這可能是因為VEGF通過破壞細胞間連接使得血管滲漏組織水腫[13],ATM激酶缺失則借由誘導VEGF的高表達影響內皮細胞屏障功能。
本研究結果表明,在高糖誘導的BRECs中,ATM激酶被激活,P-ATM表達增高并作為氧化應激中的一種保護性因子發揮作用。對這一因子加以抑制則造成細胞氧化應激狀態加劇、細胞凋亡增加、細胞間屏障功能損害。本研究初步探討了高糖環境中ATM激酶缺失誘導細胞凋亡的可能機制及其和VEGF的關系,為未來其可能作為DR的潛在治療靶點開展了一些基礎性工作。但本研究未從正面對ATM激酶的保護作用機制進行探究,是一不足。下一階段將嘗試構建ATM激酶過表達的細胞模型,同時在糖尿病動物模型中開展在體實驗,全面觀察ATM激酶在DR中所起的作用。
氧化應激與視網膜組織的炎癥反應、神經血管損害、細胞凋亡、線粒體功能紊亂等聯系密切,在糖尿病視網膜病變(DR)的病理過程中起重要作用[1-3]。共濟失調毛細血管擴張突變基因(ATM)激酶作為一種DNA損傷修復過程中的經典蛋白分子,在氧化應激中被激活并起到調控氧化應激的作用[4]。ATM缺失導致缺氧誘導因子1(HIF-1)大量增加從而加劇細胞氧化應激[5]。但目前有關ATM激酶在DR發病機制中的確切作用還不明確。為此,我們觀察高糖培養狀態下體外培養的牛視網膜微血管內皮細胞(BRECs)中ATM激酶的活性和表達變化及其對氧化應激的調節作用,初步探究ATM激酶在DR中的作用機制,為防治DR提供新的思路。現將結果報道如下。
1 材料和方法
1.1 BRECs培養及實驗分組
參照文獻[5, 6]的選擇性培養方法培養BRECs。取新鮮獲得的牛眼球,取出并剪碎視網膜微血管,加入含0.05%的膠原酶和0.025%的牛血清蛋白消化液,37℃消化45 min。應用網孔直徑88 μm的不銹鋼篩網過濾,D-hank液沖洗濾網上的組織,在離心后的小丸狀物中加入含有10%滅活胎牛血清、100 μg/ml肝素、15 μg/ml內皮細胞生長添加物、10 mmol/L羥乙基哌嗪乙磺酸的Dulbecco改良Eagle培養基,接種于包被有明膠的培養皿培養。傳代后,取第1代細胞,通過vonWillebrand因子抗體,免疫細胞化學方法對內皮細胞進行鑒定。取4~8代的細胞,傳代培養24 h后分為正常糖組、高糖組、干預組進行實驗。分別采用含5 mmol/L葡萄糖、30 mmol/L葡萄糖、30 mmol/L葡萄糖和10 μmol/L ATM激酶特異性抑制劑KU-55933的細胞培養液繼續培養48 h進行相關實驗。
1.2 實驗方法及檢測指標
細胞培養后48 h,采用蛋白免疫印跡法(Western blot)檢測細胞中ATM、磷酸化ATM(P-ATM)、細胞分裂素活化蛋白激酶(P38)、磷酸化P38(P-P38)、細胞外信號調節激酶(ERK)1/2、磷酸化ERK1/2(P-ERK1/2)的表達。應用二喹啉甲酸蛋白定量法測定裂解液提取各組細胞總蛋白。按照蛋白定量結果,以4:1的比例混合蛋白樣品及5倍上樣緩沖液,100℃加熱3~5 min,按每孔20 μg蛋白量上樣,進行電泳、轉膜、封閉、孵育一抗與二抗,包括單克隆兔抗β微管蛋白抗體、多克隆兔抗P38抗體、多克隆兔抗P-P38抗體、多克隆兔抗ERK1/2抗體、多克隆兔抗P-ERK1/2抗體(美國CST公司),辣根過氧化物酶羊抗兔IgG二抗(美國Bioworld公司),單克隆小鼠抗ATM抗體、單克隆小鼠抗P-ATM抗體(英國Abcam公司)及ATM激酶抑制劑KU55933(美國R & D公司),最后顯影定影。采用Image J圖像處理軟件進行灰度分析,以磷酸化蛋白與其總蛋白的比值表示相應蛋白的活性水平。
細胞培養后48 h,采用酶聯免疫吸附試驗(ELISA)檢測細胞上清液中血管內皮生長因子(VEGF)的含量。收集各組細胞上清液,每孔100 μl,設復孔,按照人VEGF ELISA試劑盒(上海依科賽公司)說明書進行操作。用酶聯免疫檢測儀在波長450 nm下測量吸光度[A,舊稱光密度(OD)]值,獲取細胞上清液中VEGF的含量。
細胞培養后48 h,采用細胞內ROS水平檢測試劑盒檢測細胞內ROS的水平。細胞培養后48 h,采用碘化丙啶(PI)和赫斯特染料(Hoechst)雙染色法檢測細胞凋亡情況。收集對數生長期細胞,以細胞密度2×105個細胞/孔接種于24孔培養板中。分別稱取10 mg PI或Hoechst溶于10 ml磷酸鹽緩沖液(PBS)中,終濃度為1 mg/ml。分裝后PI 4℃避光儲存,Hoechst 20℃避光儲存。配制工作液,PI:Hoechst:PBS體積比為1:1:100。吸去上清液,每孔加入工作液500 μl,置于培養箱避光培養15 min。采用熒光顯微鏡觀察細胞,以PI著紅色細胞為壞死及凋亡晚期細胞,Hoechst著藍色細胞為全部細胞,以每100個細胞中著紅色細胞數/著藍色細胞數之比為細胞凋亡率[5]。
細胞培養后48 h,終止培養,原位裝載探針,采用異硫氰酸熒光素標記的葡聚糖檢測內皮細胞屏障的通透性。按照1:1000的比例,用無血清培養液稀釋2′, 7′-二氯熒光二乙酸鹽(DCFH-DA),使終濃度為10 μmol/L。去除細胞培養液,用無血清培養液洗滌1次,加入適當體積無血清培養基稀釋好的DCFH-DA,加入的體積以能充分蓋住細胞為準。37℃細胞培養箱內孵育25~30 min。PBS洗滌細胞3次,充分去除未進入細胞內的DCFH-DA。應用熒光顯微鏡觀察并拍照。采用Image J圖像處理軟件進行定量分析。將BRECs接種于Transwell微孔膜上(美國Costar公司),微孔膜直徑24 mm,孔徑0.4 μm,參照文獻[7]的方法計算處理各組細胞旁通透率。
1.3 統計學方法
采用SPSS 17.0統計學軟件進行統計學分析。計量資料數據以均數±標準差(
2 結果
Western blot檢測結果顯示,正常糖組、高糖組及干預組P-ATM、P-P38、P-ERK1/2蛋白表達比較,差異均有統計學意義(F=436.00、14 010.00、985.10,P<0.05)。與正常糖組比較,高糖組P-ATM、P-P38、P-ERK1/2蛋白表達增高,差異均有統計學意義(t=-9.74、-4.70、-20.33,P<0.05)。與高糖組比較,干預組P-ATM蛋白表達降低,差異有統計學意義(t=5.44,P<0.01);p-p38、p-ERK1/2蛋白表達明顯增加,差異均有統計學意義(t=-18.37、-12.97,P<0.01)(圖 1)。
圖1
各組P-ATM、P-P38及P-ERK1/2蛋白表達比較。*高糖組與正常糖組比較,P<0.05;**干預組與高糖組比較,P<0.05;***干預組與高糖組比較,P<0.05??圖 2??各組BRECs中ROS含量比較。*高糖組與正常糖組比較,P<0.05;**干預組與高糖組比較,P<0.05??圖 3??各組BRECs細胞凋亡率比較。*高糖組與正常糖組比較,P<0.05;**干預組與高糖組比較,P<0.05
ELISA檢測結果顯示,正常糖組、高糖組、干預組細胞上清液中VEGF含量分別為(138.0±4.6)、(210.0±6.5)、(305.0±7.1) pg/ml,3組間差異有統計學意義(F=278.00,P<0.05)。與正常糖組比較,高糖組細胞上清液中VEGF含量有所增加,差異有統計學意義(t=-13.90,P<0.05)。與高糖組比較,干預組細胞上清液中VEGF含量明顯增加,差異也有統計學意義(t=-32.79,P<0.05)。
ROS含量檢測結果顯示,與正常糖組比較,高糖組ROS含量增高約1.5倍,差異有統計學意義(t=2.89,P<0.05)。與高糖組比較,干預組ROS含量增高約3.0倍,差異也有統計學意義(t=10.91,P<0.05)(圖 2)。
PI和Hoechst雙染色法檢測結果顯示,高糖組BRECs細胞凋亡率較正常糖組明顯增加,而又明顯低于干預組,差異均有統計學意義(t=4.31、11.14,P<0.05)。3組間BRECs細胞凋亡率比較,差異有統計學意義(F=55.51,P<0.05)(圖 3)。
正常糖組、高糖組、干預組細胞旁通透率分別為(1.12±0.13)×10-6、(1.45±0.17)×10-6、(1.66±0.12)×10-6 cm/s。與正常糖組比較,高糖組、干預組細胞旁通透率均升高,以干預組升高更明顯。3組間細胞旁通透率比較,差異有統計學意義(F=2 223.00,P<0.05)。
3 討論
本研究選取體外培養的BRECs,分別以正常濃度葡萄糖、高濃度葡萄糖以及高濃度葡萄糖聯合ATM激酶抑制劑處理細胞,觀察在不同葡萄糖濃度環境中ATM激酶的激活和表達情況、細胞內氧化應激水平、細胞凋亡水平以及細胞旁通透率的變化。結果表明,高糖組BRECs的氧化應激水平明顯比正常糖組提高,ROS激增,P-ATM激酶表達增加。說明ATM激酶在高糖環境中因為氧化應激而被ROS激活,與國外研究結果相似[4]。而在采用高糖加KU55933的干預組中,我們發現細胞的氧化應激水平與高糖組相比有進一步的提高。表明ATM激酶在氧化應激中可能起到保護作用。我們還發現,高糖組中p-p38、p-ERK1/2表達輕度增高,而干預組中p-p38、p-ERK1/2表達則大大增加。P38和ERK通路是重要的凋亡通路,同時其與細胞內氧化應激狀態聯系密切。Lu等[8]研究發現,由水銀所導致的神經元損傷中氧化應激介導的p38、ERK1/2激活是主要的細胞凋亡途徑。Tian等[9]在研究牛蒡根的神經保護作用中也發現,通過降低ROS可以抑制p38、ERK1/2的激活及其所誘導的細胞凋亡。國外亦有相關文獻報道,在ATM基因敲除鼠的神經干細胞和星形膠質細胞中P38和ERK通路的激活,通過上調CDK抑制物和凋亡相關基因的表達,抑制了細胞的增生,誘導細胞生長停滯和凋亡[10, 11]。本研究結果同樣證實了ATM激酶的抑制導致P38和ERK通路的激活,從而增加了細胞的凋亡。據此我們推測,在BRECs中,ATM激酶缺失后,細胞內氧化應激失控,ROS爆發式產生,進而激活P38和ERK通路造成BRECs凋亡。說明ATM激酶在氧化應激中至關重要,其活性關系到視網膜血管內皮細胞凋亡與否。在DR病理變化中如能保持和提高ATM激酶的活性,控制細胞氧化應激,可能對控制DR發展起到積極作用。
高糖誘導的VEGF增高和由于高水平VEGF所導致的血視網膜屏障破壞、血管滲漏以及后期的新生血管形成都與DR病理過程密切相關。我們在探索ATM與VEGF的關系時發現,高糖組VEGF表達高于正常糖組,而干預組VEGF水平遠遠高于高糖組。我們分析認為,高糖環境下細胞處于氧化應激的狀態并產生大量ROS,其原因則是細胞能量代謝系統功能紊亂,線粒體電子傳遞鏈破壞,此時細胞無法正常利用氧進行有氧呼吸,處于一種缺氧的狀態[12]。ATM激酶作為一種對于缺氧和氧化應激的應答,起到保護性作用。當ATM激酶被抑制或敲除,細胞缺氧,氧化應激水平將進一步提高,導致VEGF上調。Ousset等[5]在敲除ATM基因的海拉細胞中發現,通過HIF-1高表達,可誘導作為其下游的VEGF表達增高。該結果與我們的推測一致。我們還發現各組細胞旁通透性測定結果與VEGF表達趨勢一致。這可能是因為VEGF通過破壞細胞間連接使得血管滲漏組織水腫[13],ATM激酶缺失則借由誘導VEGF的高表達影響內皮細胞屏障功能。
本研究結果表明,在高糖誘導的BRECs中,ATM激酶被激活,P-ATM表達增高并作為氧化應激中的一種保護性因子發揮作用。對這一因子加以抑制則造成細胞氧化應激狀態加劇、細胞凋亡增加、細胞間屏障功能損害。本研究初步探討了高糖環境中ATM激酶缺失誘導細胞凋亡的可能機制及其和VEGF的關系,為未來其可能作為DR的潛在治療靶點開展了一些基礎性工作。但本研究未從正面對ATM激酶的保護作用機制進行探究,是一不足。下一階段將嘗試構建ATM激酶過表達的細胞模型,同時在糖尿病動物模型中開展在體實驗,全面觀察ATM激酶在DR中所起的作用。