Gordon綜合征是一種罕見的單基因高血壓疾病,發病率低,臨床表現異質性大,目前對該疾病的基因診斷報道較少。本文報道1例賴氨酸缺乏蛋白激酶1基因新的突變位點[c.3029G>A(p.Gly1010Glu)]致Gordon綜合征患者,女,21歲。患者體檢時發現血壓升高,噻嗪類利尿劑治療后效果顯著,明確診斷為Gordon綜合征,此次病例拓展了Gordon綜合征的突變譜,為其不同的臨床表現提供依據。本文旨在與臨床醫生共同學習探討,提高對單基因高血壓的認識。
臨床資料 患者,女,21歲,因“發現血壓升高8個月”于我院就診。8個月前因體檢發現血壓升高(血壓值不詳),在家監測血壓最高為215/155 mm Hg(1 mm Hg=0.133 kPa),不伴頭昏、頭痛、視物模糊、心慌、胸痛、腰痛、血尿、夜尿增多、乏力、四肢麻木、打鼾等,平素不規律監測血壓(血壓值不詳)。后于我院門診就診,監測血壓波動于205~215/146~155 mmHg,否認高血壓家族史。查體:心率126次/min,血壓215/151 mm Hg,身高163 cm,體重51 kg,體重指數(body mass index,BMI)19.20 kg/m2。心前區無隆起或凹陷,心尖搏動點位于左側第5肋間鎖骨中線內約1.00 cm,心界不大,心律齊,未聞及心音分裂及額外心音,各瓣膜區未聞及雜音,未聞及血管雜音,雙下肢無水腫。
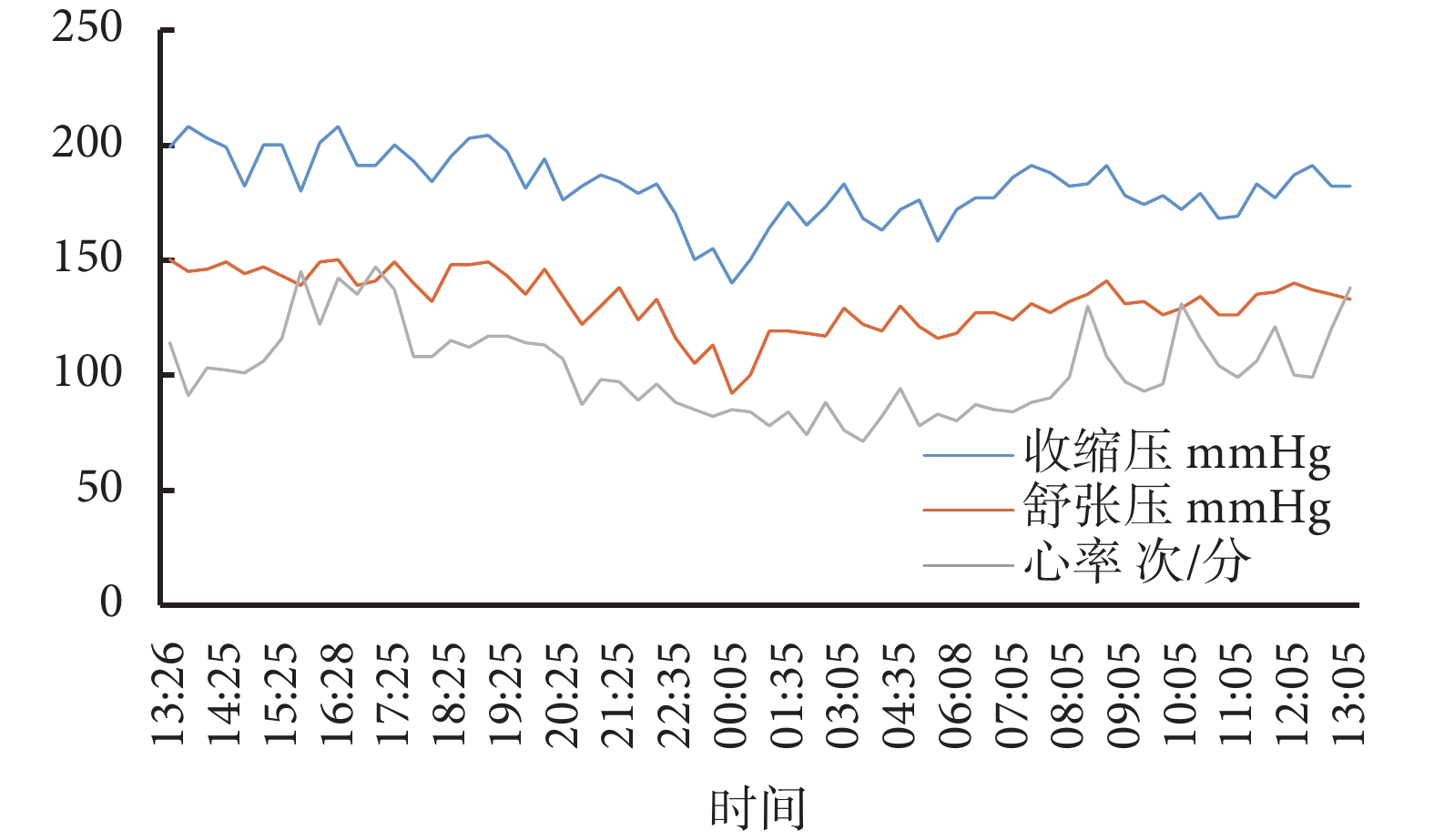
入院后完善輔助檢查,血鉀2.79 mmol/L,血氯95.30 mmol/L,血肌酐61 μmol/L,估算腎小球濾過率125.72 mL/(min·1.73m2);24 h尿電解質:鉀33.97 mmol/24h(參考值40~80 mmol/24h);小便常規:尿蛋白定性1+,尿白蛋白/肌酐25.30 mg/g;血漿腎素活性(立位)>12 ng/(mL·h)[參考值0.93~6.56 ng/(mL·h)],血管緊張素Ⅱ(立位) 798.87 ng/L(參考值55.30~115.30 ng/L),醛固酮(立位)46.12 ng/dL(參考值9.80~27.50 ng/dL,1 ng/dL=27.70 pmol/L),血去甲腎上腺素872 ng/L(參考值174.00~357.00 ng/L),腎上腺素70 ng/L(參考值60.00~104.00 ng/L),促腎上腺皮質激素15.87 ng/L,血皮質醇(8點)484 nmol/L;超聲心動圖、腎及腎動脈彩色超聲、頸動脈彩色超聲、腎及腎上腺磁共振增強掃描未見異常。動態血壓:24 h平均血壓180/130 mm Hg,白晝平均血壓187/137 mm Hg,夜晚平均血壓166/117 mm Hg;見圖1。
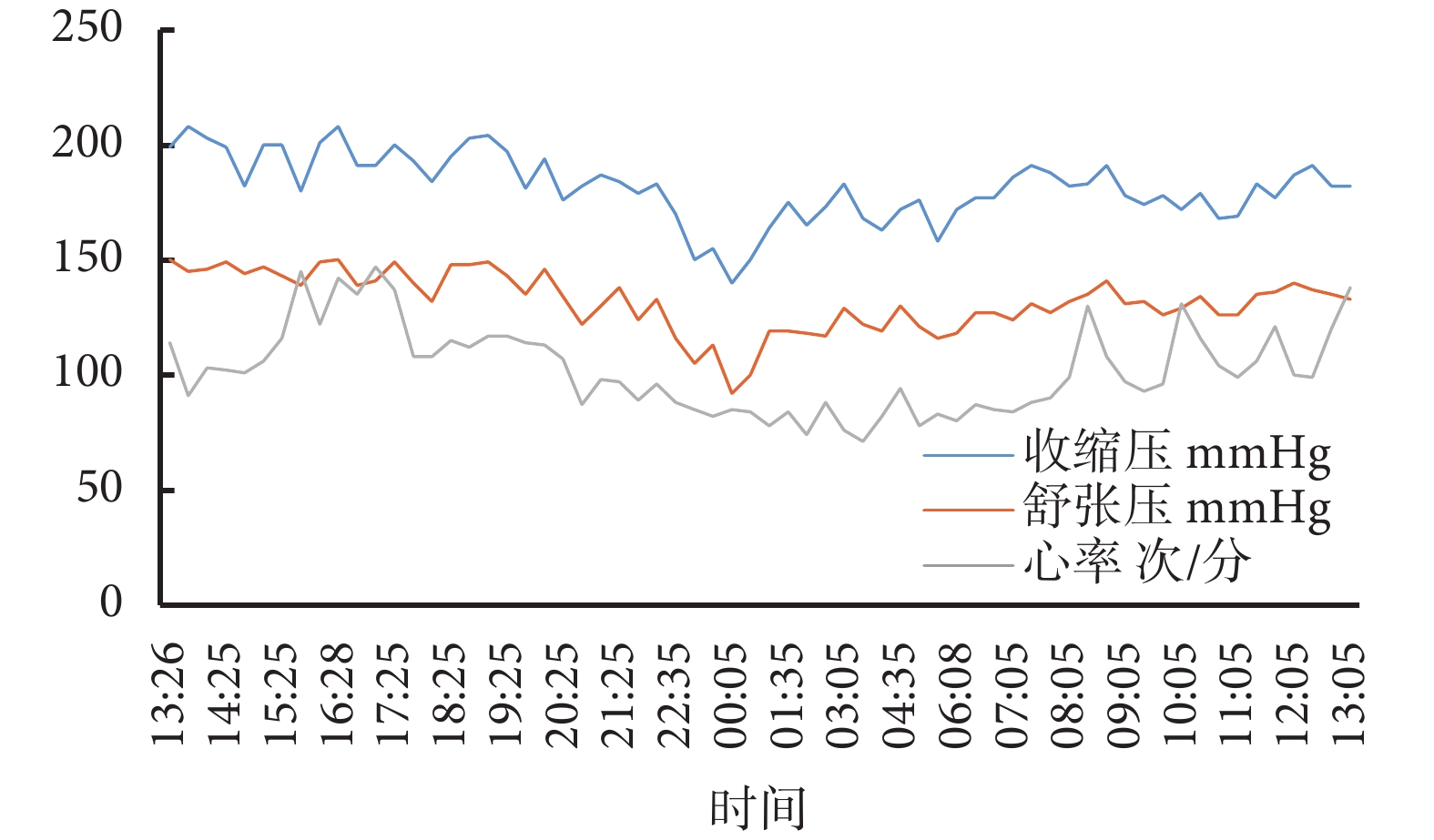 圖1
用藥前動態血壓和心率監測
圖1
用藥前動態血壓和心率監測
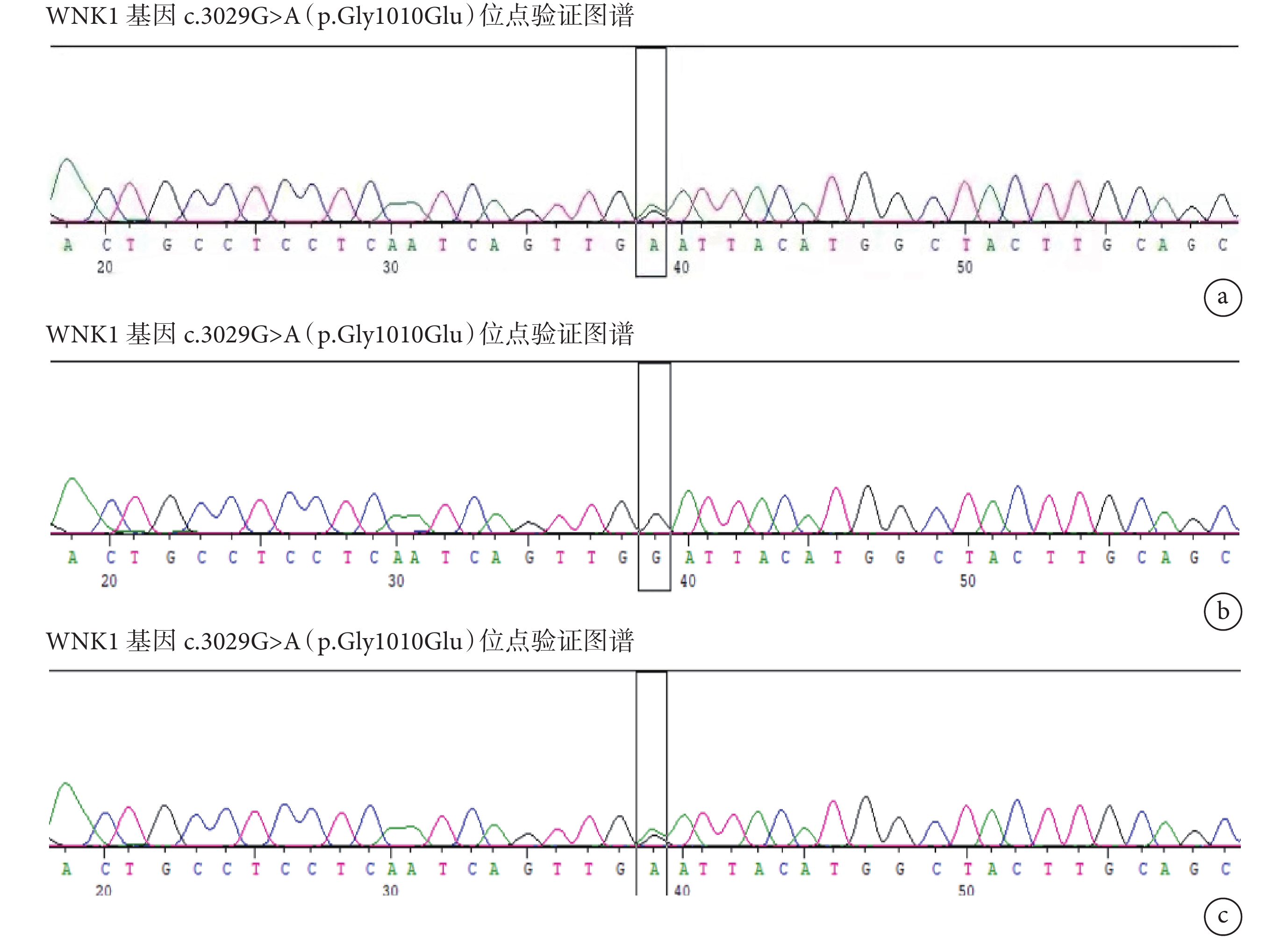
排除嗜鉻細胞瘤、腎動脈狹窄、原發性醛固酮增多癥等常見繼發性高血壓可能。給予生活方式指導,予硝苯地平控釋片30 mg 1次/d控制血壓,血壓波動于168~182/116~134 mm Hg。行高血壓基因檢測,發現患者攜帶賴氨酸缺乏蛋白激酶1(WNK1)基因c.3029G>A(p.Gly1010Glu)雜合異位點;見圖2、表1。初步考慮為Gordon綜合征,診斷性使用氫氯噻嗪25 mg 1次/d治療。
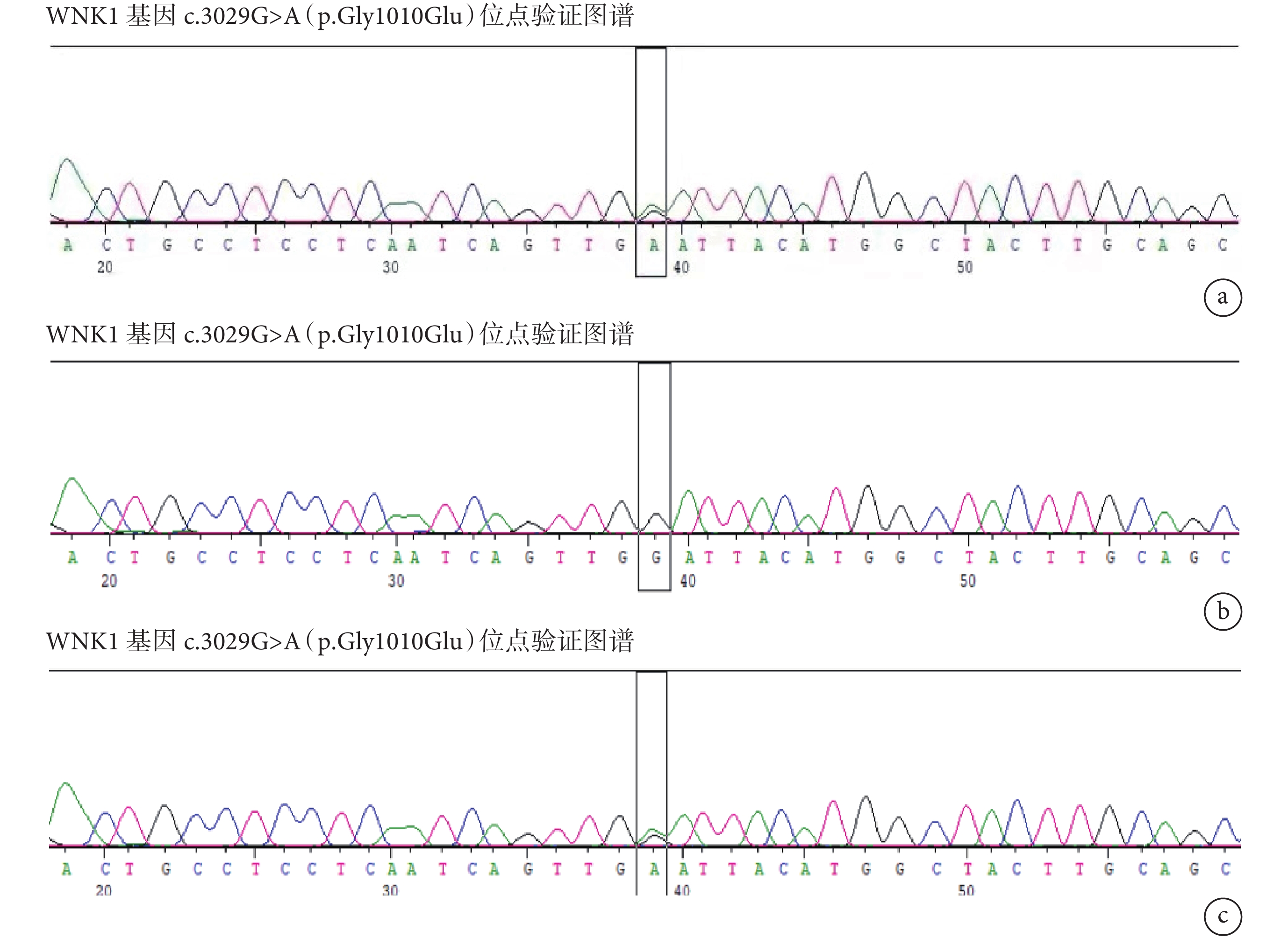 圖2
高血壓基因結果
圖2
高血壓基因結果
a:先證者基因圖譜;b:先證者父親基因圖譜;c:先證者母親基因圖譜
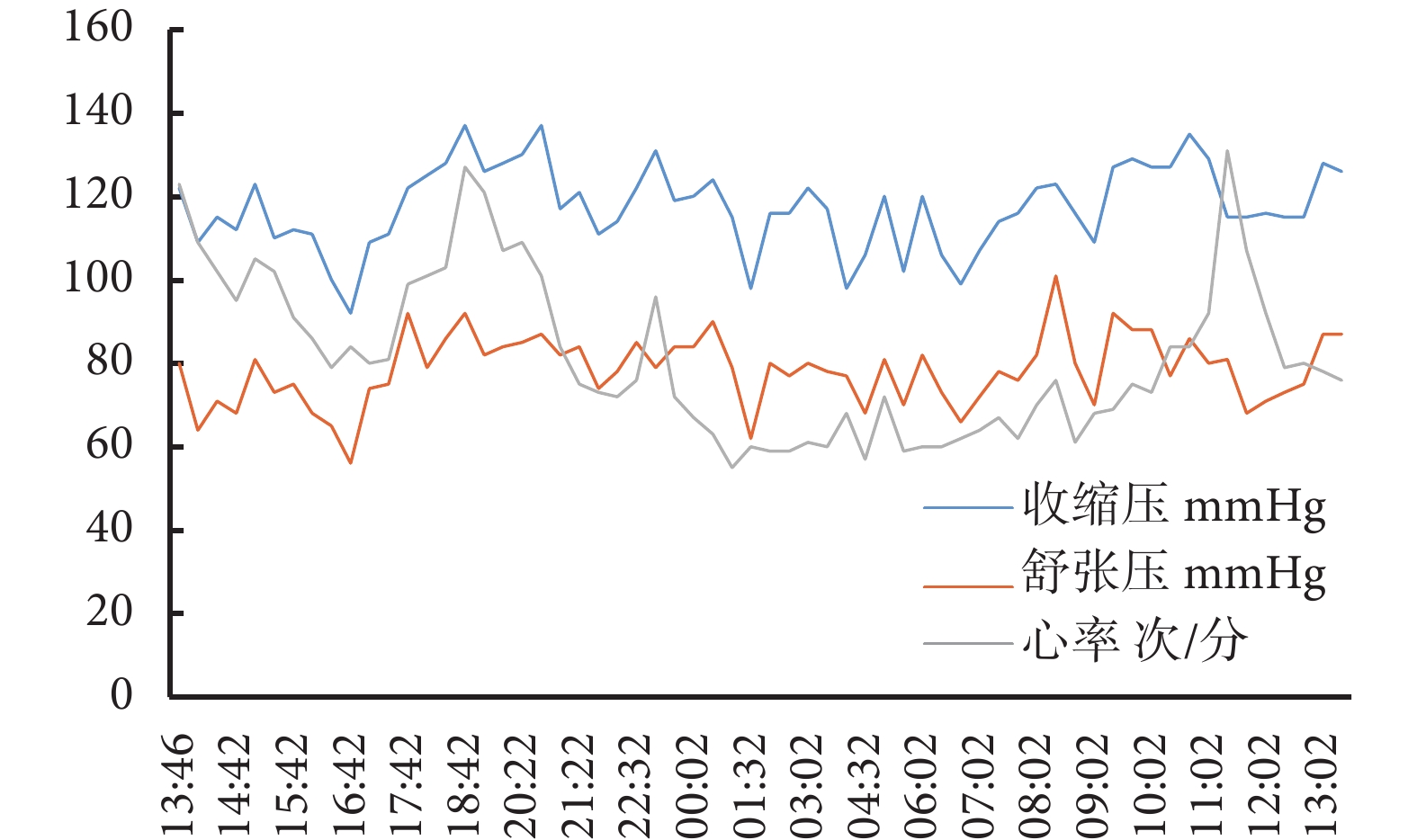
患者使用氫氯噻嗪25 mg,1次/d,血壓較前明顯降低,波動于110~130/70~90 mm Hg。1年后門診復查24 h動態血壓:24 h平均血壓118/79 mm Hg,白晝平均血壓119/79 mm Hg,夜晚平均血壓115/79 mm Hg;見圖3。血鉀3.98 mmol/L,血氯101.90 mmol/L,血肌酐49 μmol/L,尿素3.90 mmol/L,估算腎小球濾過率133.23 mL/(min·1.73m2),尿蛋白定性(?)。后患者在家自行監測血壓,1個月后門診復診,因季節變換,血壓波動于110~140/90~100 mm Hg,調整藥物為厄貝沙坦氫氯噻嗪片1片,1次/d。規律隨訪患者,血壓波動于108~130/70~90 mm Hg,繼續予以厄貝沙坦氫氯噻嗪片1片,1次/d治療及生活方式管理。
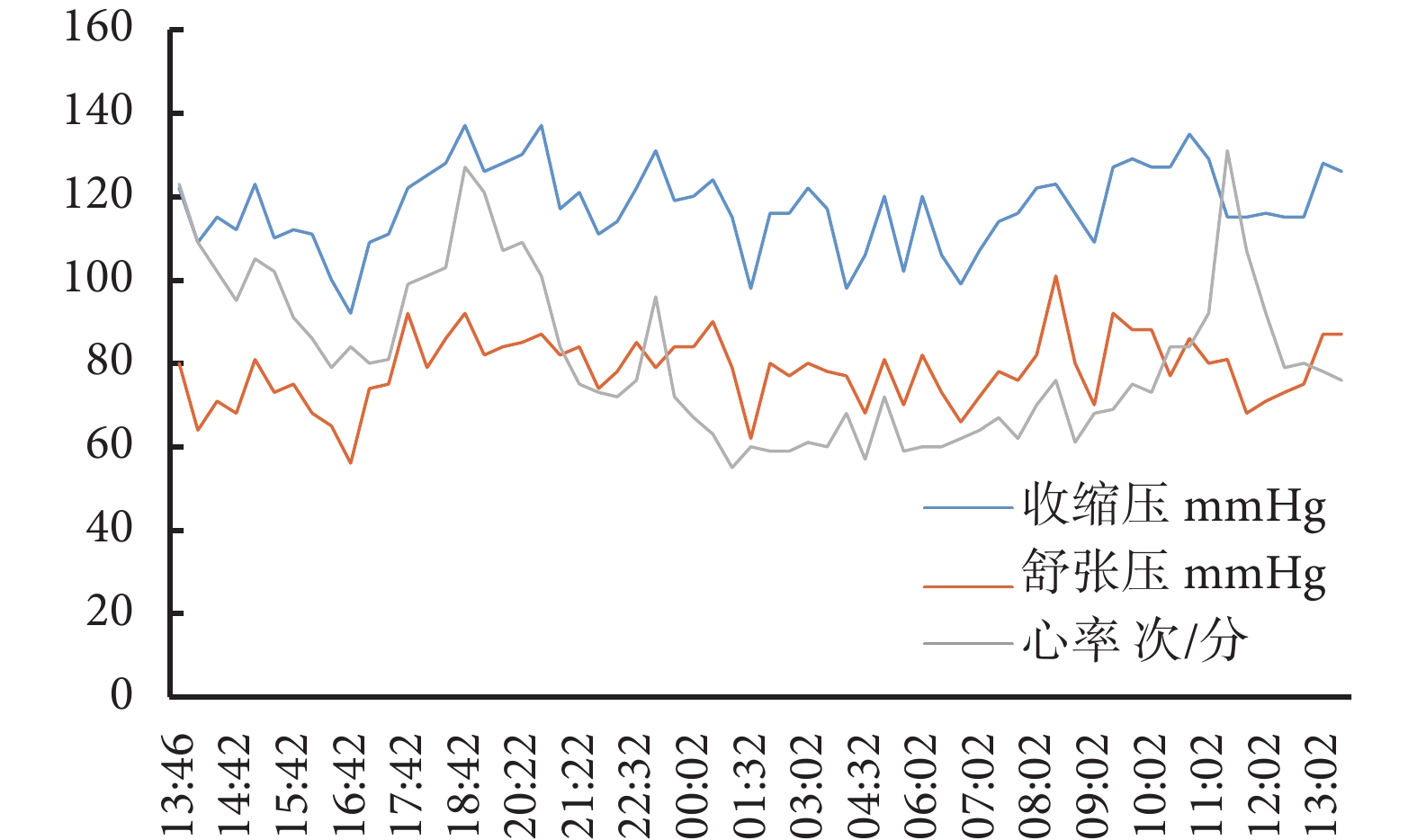 圖3
用藥后動態血壓監測
圖3
用藥后動態血壓監測
本研究經四川大學華西醫院生物醫學倫理委員會審批,批準號:2020年審(261)號。
討論 目前青年高血壓逐漸增多[1],發病率已經達到 0%[2],但知曉率、治療率和控制率遠低于中老年高血壓[3]。高血壓中繼發性高血壓約占5%~15%[4]。單基因遺傳性高血壓是指由1個基因突變引起的高血壓,常符合孟德爾遺傳規律[5]。單基因遺傳性高血壓亦屬于繼發性高血壓,由于認識不足、基因檢測缺乏等限制,常被忽視,但其患病率并不低[6],全國2000萬~3500萬高血壓患者可能是單基因高血壓[7]。現已明確的單基因致病型高血壓有17種[8],是臨床診治的難點。
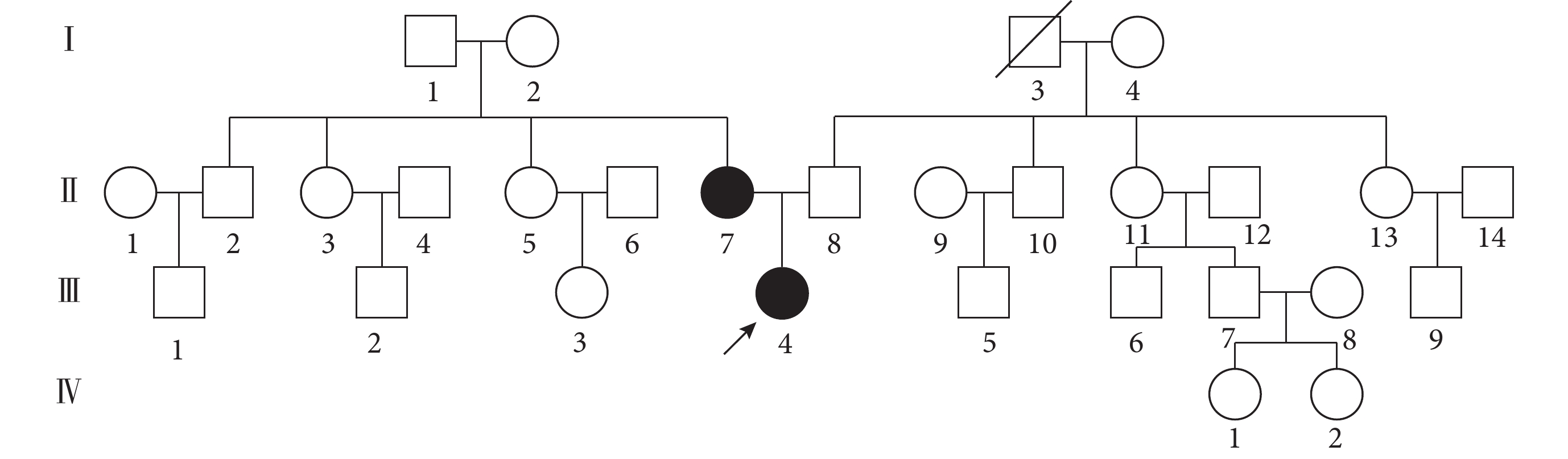
Gordon綜合征為單基因遺傳性高血壓之一,典型的臨床特征是高血壓和高鉀血癥,目前發現其發病機制與WNK1、WNK4,克爾希樣蛋白3(KLHL3)和泛素連接酶3(CUL3)基因相關[9]。本例患者為青年女性,血壓達210/150 mm Hg以上,排除常見繼發性高血壓可能,進一步完善高血壓基因檢測,發現患者攜帶WNK1基因c.3029G>A(p.Gly1010Glu)雜合異位點。嚴格遵循知情同意原則,對患者父母進行高血壓基因家系驗證:患者攜帶的WNK1基因c.3029G>A(p.Gly1010Glu)遺傳自健康正常母親(圖2),反復詢問患者家族史,否認直系親屬高血壓病史(圖4)。該基因突變與Gordon綜合征相關,初步考慮為Gordon綜合征。
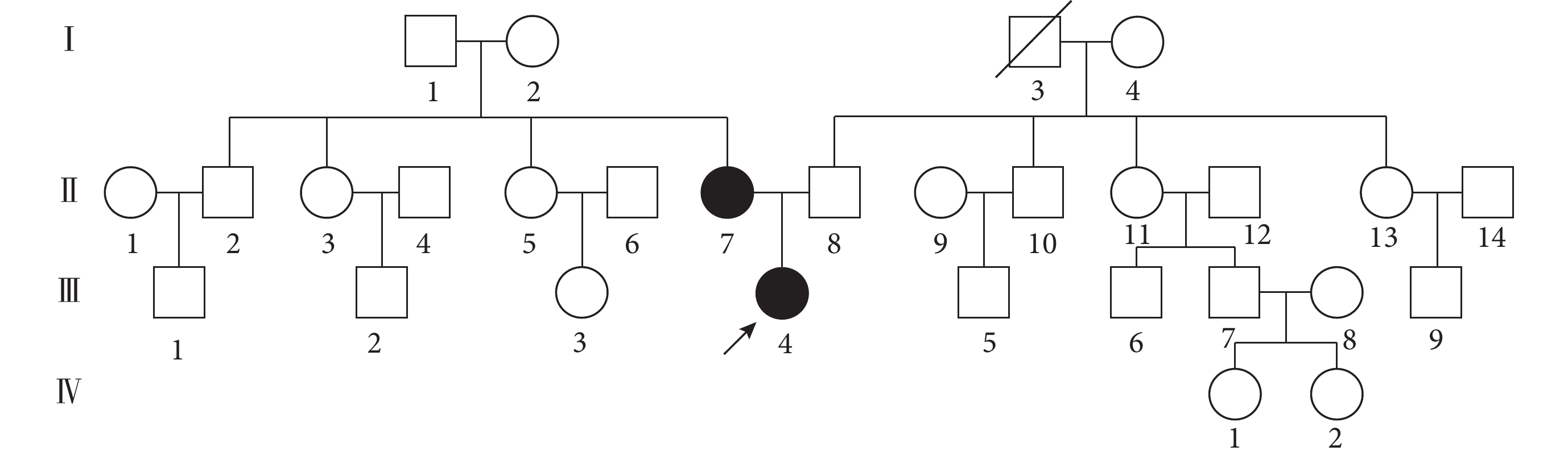 圖4
Gordon綜合征患者家系圖
圖4
Gordon綜合征患者家系圖
注:黑色填充示意Gordon相關基因攜帶者;斜線示意死亡;箭頭示意先證者;正方形代表男性,圓形代表女性
Gordon綜合征報道少,迄今為止世界范圍內僅報道Gordon綜合征約180余例[10],臨床表現異質性大,診斷難度高。本文報道的WNK1基因新突變位點(c.3029G>A(p.Gly1010Glu)致Gordon綜合征病例既往未見報道。該病例表現為高血壓,合并低鉀血癥、血漿腎素活性及醛固酮升高,與既往報道Gordon綜合征病例表現不同,具有獨特性。為比較本例發現的WNK1基因新突變位點與既往報道過的基因突變位點所致臨床表現的異同,對Gordon綜合征相關知識進行文獻復習。Gordon綜合征的致病突變于2001年被第1次鑒定[11],以Pseudohypoaldosteronism type Ⅱ、Familial hyperkalemia、Gordon syndrome、Gordon綜合征、假性醛固酮減少癥2型、家族性高血鉀性高血壓為關鍵詞,檢索PubMed、知網和萬方數據庫。納入2001年1月—2021年7月在中英文文獻中公開報道過的經基因檢測確診的Gordon綜合征文獻15篇[12-26],發現突變基因20種,案例41例。其中高血壓27例(65.9%)、高鉀血癥33例(80.5%)、低醛固酮血癥6例(14.6%)、低腎素9例(22.0%)、代謝性酸中毒20例(48.8%)、相同遺傳基因12例(29.3%)、高血壓家族史18例(43.9%),證實了Gordon綜合征與WNK1、WNK4、KLHL3和CUL3基因相關,典型臨床特征是高血壓和高鉀血癥。
回顧既往報道的41例攜帶Gordon綜合征基因患者,其中高血壓合并高鉀血癥23例(56.1%),大多數案例與Gordon綜合征典型癥狀相符,但仍有少部分表現獨特,僅憑臨床特征難以確診,體現了基因檢測的重要性。本例患者表現為高血壓合并低鉀血癥、血漿腎素活性及醛固酮升高,無高血壓家族史,基因僅來自于突變位點相同的健康母親。與既往報道的Gordon綜合征相比,本例患者致病基因在既往文獻中從未公開報道,且臨床表現與既往案例不同,具有獨特性,故分享病例,旨在臨床醫生共同學習探討,為Gordon綜合征更多的基因突變位點及臨床表現提供依據。
Gordon綜合征發病的核心機制是WNK1或WNK4基因缺陷,導致NaCl協同轉運蛋白過度激活[11],腎的NaCl重吸收增加,進而顯著升高血壓[27]。由于遠端腎單位的NaCl轉運蛋白對噻嗪類利尿劑敏感,因此噻嗪類利尿劑是Gordon綜合征患者控制血壓的首選藥物[28]。本案例給予患者氫氯噻嗪類藥物,聯合低鈉飲食及運動干預,血壓控制效果顯著,明確診斷Gordon綜合征。考慮長期服用噻嗪類利尿劑對腎功能的影響,每3個月監測患者血尿電解質及腎功能[10]。此外,該患者自行規律測量血壓和隨診,整體依從性較高。
綜上所述,本研究發現了WNK1基因的一個新突變位點(c.3029G>A(p.Gly1010Glu),拓寬了Gordon綜合征基因譜;Gordon綜合征臨床表現異質性大,可表現為高血壓和低鉀血癥,本研究為其不同的臨床表現提供了依據。基因檢查目前已成為臨床診斷重要的助力,因此我們在臨床工作中應提高對單基因疾病的認識,面對難治性青年高血壓患者,排除常見高血壓繼發因素后,應當進行單基因高血壓疾病的致病基因篩查,協助臨床診斷及干預治療。
利益沖突:無。
作者貢獻:陳欣收集資料,撰寫文章;陳欣、方湘、賈禹整理與分析數據,設計論文;廖曉陽審閱與修改論文。
臨床資料 患者,女,21歲,因“發現血壓升高8個月”于我院就診。8個月前因體檢發現血壓升高(血壓值不詳),在家監測血壓最高為215/155 mm Hg(1 mm Hg=0.133 kPa),不伴頭昏、頭痛、視物模糊、心慌、胸痛、腰痛、血尿、夜尿增多、乏力、四肢麻木、打鼾等,平素不規律監測血壓(血壓值不詳)。后于我院門診就診,監測血壓波動于205~215/146~155 mmHg,否認高血壓家族史。查體:心率126次/min,血壓215/151 mm Hg,身高163 cm,體重51 kg,體重指數(body mass index,BMI)19.20 kg/m2。心前區無隆起或凹陷,心尖搏動點位于左側第5肋間鎖骨中線內約1.00 cm,心界不大,心律齊,未聞及心音分裂及額外心音,各瓣膜區未聞及雜音,未聞及血管雜音,雙下肢無水腫。
入院后完善輔助檢查,血鉀2.79 mmol/L,血氯95.30 mmol/L,血肌酐61 μmol/L,估算腎小球濾過率125.72 mL/(min·1.73m2);24 h尿電解質:鉀33.97 mmol/24h(參考值40~80 mmol/24h);小便常規:尿蛋白定性1+,尿白蛋白/肌酐25.30 mg/g;血漿腎素活性(立位)>12 ng/(mL·h)[參考值0.93~6.56 ng/(mL·h)],血管緊張素Ⅱ(立位) 798.87 ng/L(參考值55.30~115.30 ng/L),醛固酮(立位)46.12 ng/dL(參考值9.80~27.50 ng/dL,1 ng/dL=27.70 pmol/L),血去甲腎上腺素872 ng/L(參考值174.00~357.00 ng/L),腎上腺素70 ng/L(參考值60.00~104.00 ng/L),促腎上腺皮質激素15.87 ng/L,血皮質醇(8點)484 nmol/L;超聲心動圖、腎及腎動脈彩色超聲、頸動脈彩色超聲、腎及腎上腺磁共振增強掃描未見異常。動態血壓:24 h平均血壓180/130 mm Hg,白晝平均血壓187/137 mm Hg,夜晚平均血壓166/117 mm Hg;見圖1。
圖1
用藥前動態血壓和心率監測
排除嗜鉻細胞瘤、腎動脈狹窄、原發性醛固酮增多癥等常見繼發性高血壓可能。給予生活方式指導,予硝苯地平控釋片30 mg 1次/d控制血壓,血壓波動于168~182/116~134 mm Hg。行高血壓基因檢測,發現患者攜帶賴氨酸缺乏蛋白激酶1(WNK1)基因c.3029G>A(p.Gly1010Glu)雜合異位點;見圖2、表1。初步考慮為Gordon綜合征,診斷性使用氫氯噻嗪25 mg 1次/d治療。
圖2
高血壓基因結果
a:先證者基因圖譜;b:先證者父親基因圖譜;c:先證者母親基因圖譜
患者使用氫氯噻嗪25 mg,1次/d,血壓較前明顯降低,波動于110~130/70~90 mm Hg。1年后門診復查24 h動態血壓:24 h平均血壓118/79 mm Hg,白晝平均血壓119/79 mm Hg,夜晚平均血壓115/79 mm Hg;見圖3。血鉀3.98 mmol/L,血氯101.90 mmol/L,血肌酐49 μmol/L,尿素3.90 mmol/L,估算腎小球濾過率133.23 mL/(min·1.73m2),尿蛋白定性(?)。后患者在家自行監測血壓,1個月后門診復診,因季節變換,血壓波動于110~140/90~100 mm Hg,調整藥物為厄貝沙坦氫氯噻嗪片1片,1次/d。規律隨訪患者,血壓波動于108~130/70~90 mm Hg,繼續予以厄貝沙坦氫氯噻嗪片1片,1次/d治療及生活方式管理。
圖3
用藥后動態血壓監測
本研究經四川大學華西醫院生物醫學倫理委員會審批,批準號:2020年審(261)號。
討論 目前青年高血壓逐漸增多[1],發病率已經達到 0%[2],但知曉率、治療率和控制率遠低于中老年高血壓[3]。高血壓中繼發性高血壓約占5%~15%[4]。單基因遺傳性高血壓是指由1個基因突變引起的高血壓,常符合孟德爾遺傳規律[5]。單基因遺傳性高血壓亦屬于繼發性高血壓,由于認識不足、基因檢測缺乏等限制,常被忽視,但其患病率并不低[6],全國2000萬~3500萬高血壓患者可能是單基因高血壓[7]。現已明確的單基因致病型高血壓有17種[8],是臨床診治的難點。
Gordon綜合征為單基因遺傳性高血壓之一,典型的臨床特征是高血壓和高鉀血癥,目前發現其發病機制與WNK1、WNK4,克爾希樣蛋白3(KLHL3)和泛素連接酶3(CUL3)基因相關[9]。本例患者為青年女性,血壓達210/150 mm Hg以上,排除常見繼發性高血壓可能,進一步完善高血壓基因檢測,發現患者攜帶WNK1基因c.3029G>A(p.Gly1010Glu)雜合異位點。嚴格遵循知情同意原則,對患者父母進行高血壓基因家系驗證:患者攜帶的WNK1基因c.3029G>A(p.Gly1010Glu)遺傳自健康正常母親(圖2),反復詢問患者家族史,否認直系親屬高血壓病史(圖4)。該基因突變與Gordon綜合征相關,初步考慮為Gordon綜合征。
圖4
Gordon綜合征患者家系圖
注:黑色填充示意Gordon相關基因攜帶者;斜線示意死亡;箭頭示意先證者;正方形代表男性,圓形代表女性
Gordon綜合征報道少,迄今為止世界范圍內僅報道Gordon綜合征約180余例[10],臨床表現異質性大,診斷難度高。本文報道的WNK1基因新突變位點(c.3029G>A(p.Gly1010Glu)致Gordon綜合征病例既往未見報道。該病例表現為高血壓,合并低鉀血癥、血漿腎素活性及醛固酮升高,與既往報道Gordon綜合征病例表現不同,具有獨特性。為比較本例發現的WNK1基因新突變位點與既往報道過的基因突變位點所致臨床表現的異同,對Gordon綜合征相關知識進行文獻復習。Gordon綜合征的致病突變于2001年被第1次鑒定[11],以Pseudohypoaldosteronism type Ⅱ、Familial hyperkalemia、Gordon syndrome、Gordon綜合征、假性醛固酮減少癥2型、家族性高血鉀性高血壓為關鍵詞,檢索PubMed、知網和萬方數據庫。納入2001年1月—2021年7月在中英文文獻中公開報道過的經基因檢測確診的Gordon綜合征文獻15篇[12-26],發現突變基因20種,案例41例。其中高血壓27例(65.9%)、高鉀血癥33例(80.5%)、低醛固酮血癥6例(14.6%)、低腎素9例(22.0%)、代謝性酸中毒20例(48.8%)、相同遺傳基因12例(29.3%)、高血壓家族史18例(43.9%),證實了Gordon綜合征與WNK1、WNK4、KLHL3和CUL3基因相關,典型臨床特征是高血壓和高鉀血癥。
回顧既往報道的41例攜帶Gordon綜合征基因患者,其中高血壓合并高鉀血癥23例(56.1%),大多數案例與Gordon綜合征典型癥狀相符,但仍有少部分表現獨特,僅憑臨床特征難以確診,體現了基因檢測的重要性。本例患者表現為高血壓合并低鉀血癥、血漿腎素活性及醛固酮升高,無高血壓家族史,基因僅來自于突變位點相同的健康母親。與既往報道的Gordon綜合征相比,本例患者致病基因在既往文獻中從未公開報道,且臨床表現與既往案例不同,具有獨特性,故分享病例,旨在臨床醫生共同學習探討,為Gordon綜合征更多的基因突變位點及臨床表現提供依據。
Gordon綜合征發病的核心機制是WNK1或WNK4基因缺陷,導致NaCl協同轉運蛋白過度激活[11],腎的NaCl重吸收增加,進而顯著升高血壓[27]。由于遠端腎單位的NaCl轉運蛋白對噻嗪類利尿劑敏感,因此噻嗪類利尿劑是Gordon綜合征患者控制血壓的首選藥物[28]。本案例給予患者氫氯噻嗪類藥物,聯合低鈉飲食及運動干預,血壓控制效果顯著,明確診斷Gordon綜合征。考慮長期服用噻嗪類利尿劑對腎功能的影響,每3個月監測患者血尿電解質及腎功能[10]。此外,該患者自行規律測量血壓和隨診,整體依從性較高。
綜上所述,本研究發現了WNK1基因的一個新突變位點(c.3029G>A(p.Gly1010Glu),拓寬了Gordon綜合征基因譜;Gordon綜合征臨床表現異質性大,可表現為高血壓和低鉀血癥,本研究為其不同的臨床表現提供了依據。基因檢查目前已成為臨床診斷重要的助力,因此我們在臨床工作中應提高對單基因疾病的認識,面對難治性青年高血壓患者,排除常見高血壓繼發因素后,應當進行單基因高血壓疾病的致病基因篩查,協助臨床診斷及干預治療。
利益沖突:無。
作者貢獻:陳欣收集資料,撰寫文章;陳欣、方湘、賈禹整理與分析數據,設計論文;廖曉陽審閱與修改論文。